
The presentation by Professor Zoe Cournia, who is at the Biomedical Research Foundation Academy of Athens in Greece, was about the power of supercomputing and novel allosteric drug discovery. Watch the full presentation video below:
Transcript of the presentation by Professor Zoe Cournia:
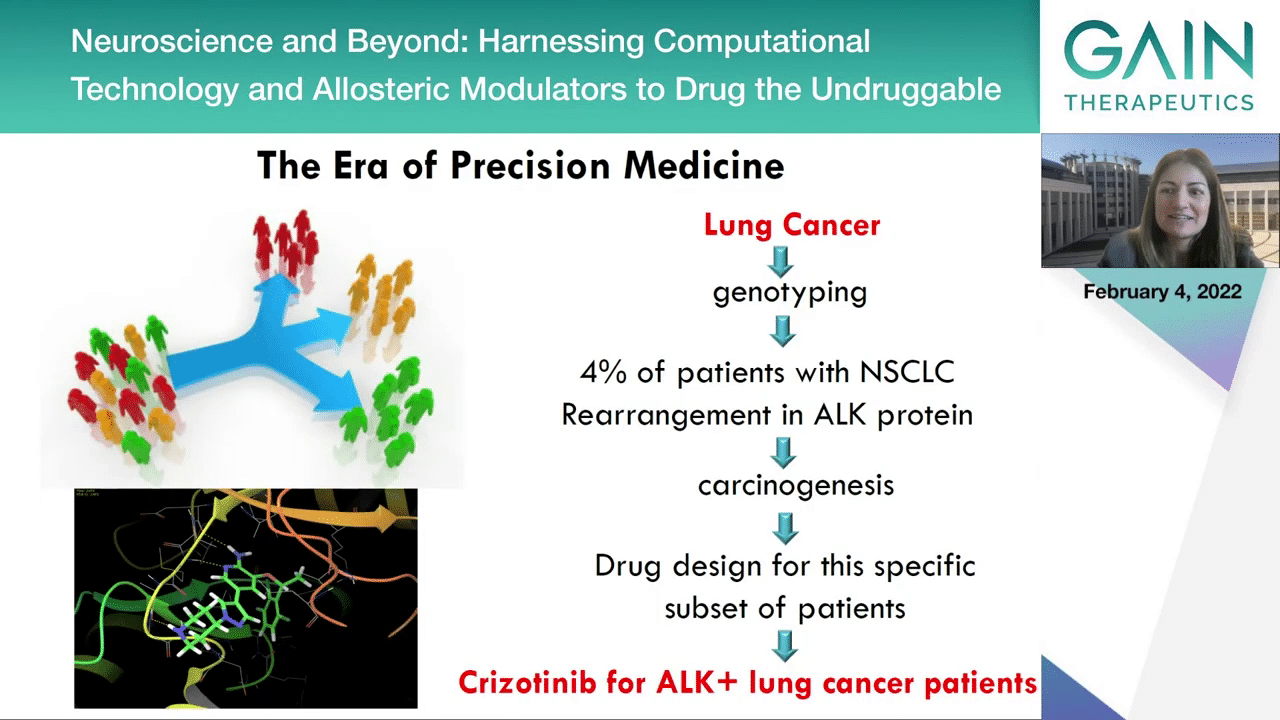
Pharmaceutical research is now framed in the context of precision medicine where patient populations are stratified and we can offer medicines according to the patient’s very own specific genotype and provide the right drug to the right patient at the right time and at the right dose, such as the example of the 4% of non-small cell lung cancer patients that have been genotyped and it was found that they all bear a rearrangement in their anaplastic lymphoma kinase protein that leads to carcinogenesis. Drug discovery for this specific subset of patients led to a revolutionary at the time, crizotinib, that can be prescribed for anaplastic lymphoma kinase patients that have this rearrangement. That was really revolutionary at the time. Next slide, please.
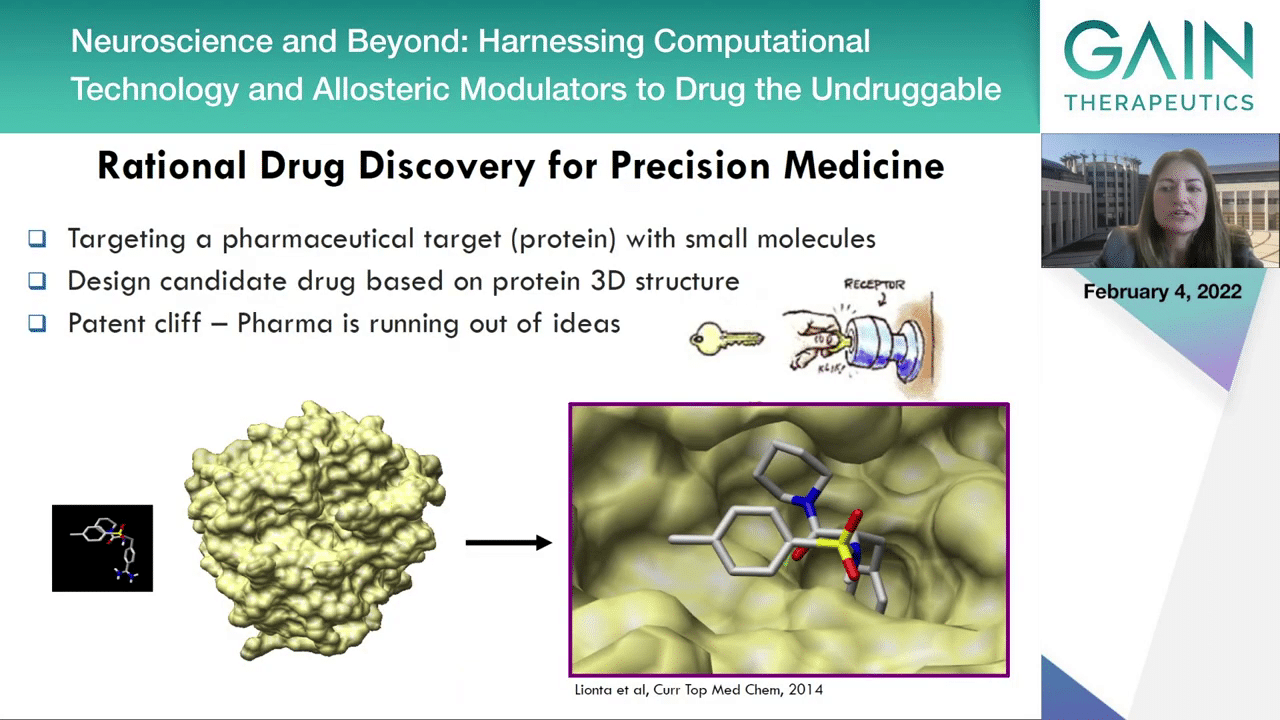
Rational drug discovery is a cornerstone for precision medicine because, for example, by knowing the 3-dimensional structural pharmaceutical target which is usually a protein, we can now design molecules specifically designed for the target of interest. However, this strategy has so far targeted the active sites of enzymes and as we are progressing through the years in pharmaceutical research we find that this technique or approach is not devoid of problems.
First of all, given a protein site it can look very similar to active sites of other family members, making it difficult to design a compound that doesn’t actually unlock all of these enzymes and cause undesirable side effects.
And second, because each enzyme has one or at most two active or steric sites, so it is really difficult to design more than one blockbluster drug for every protein, leading the pharma industry to a patent cliff.
What could be done to produce more ideas? Next slide.
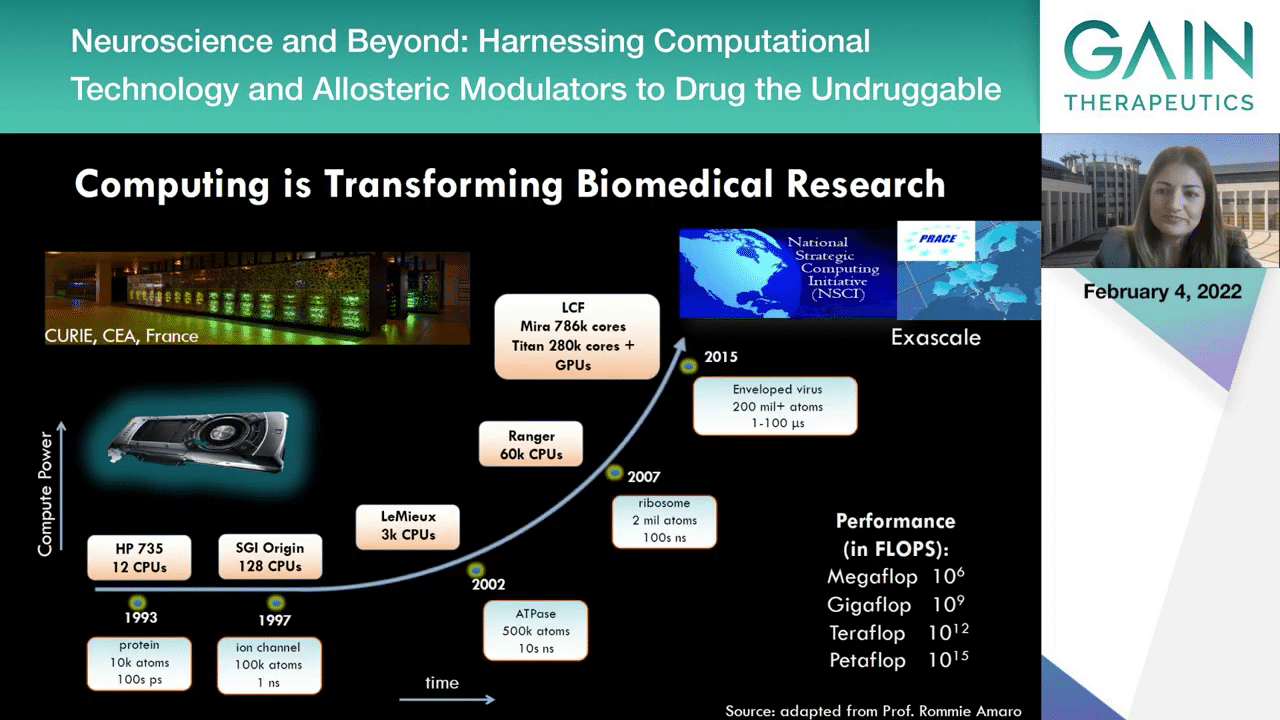
We can redefine, actually, drug discovery by using revolutionary technologies to discover new binding pockets and predict their druggability. One of the technologies that are currently transforming biomedical research is high performance computing. With high performance computing we can now reach unprecedented length and time scales for understanding the structure and dynamics of biomolecules, enabling the generation of new therapeutic strategies. Actually, you can see in this slide that in 2015 an enveloped virus to 200 million atoms was already simulating and we are now leading to even more powerful computing days with the Exascale era just on the verge.

On the next slide, how can we use this technology to actually rediscover all targets with new drugs, but also use this technology to drug proteins that have been considered so far undruggable? Next slide.

The answer is a phenomenon that was first observed in 1904 by Bohr in hemoglobin termed allostery, which in Greek means a different site. Thus, we now know that enzymes are not only regulated simply by their active or orthosteric sites, but also by other secondary sites that we have not yet discovered, at least not for all of the proteins. That’s why allostery has been termed as the second secret of life. Next slide.

But what is allostery exactly? Allostery is a phenomenon when a ligand or a modulator binds to a site other than the orthosteric or active site, changing the confirmation of the orthosteric site. The substrate can still bind but no catalysis can happen because the allosteric inhibitor or modulator has actually changed the confirmation of the active site and the substrate can no longer do the catalysis. Next slide.

It’s really amazing how as I mentioned it was discovered in 1904, so we only know this phenomenon for 120 years, and with the first allosteric drug approval by the FDA happening in 2004, just 20 years ago, you can understand how this opens up a completely new drug design strategy. Next slide.
Let’s see some real life examples.
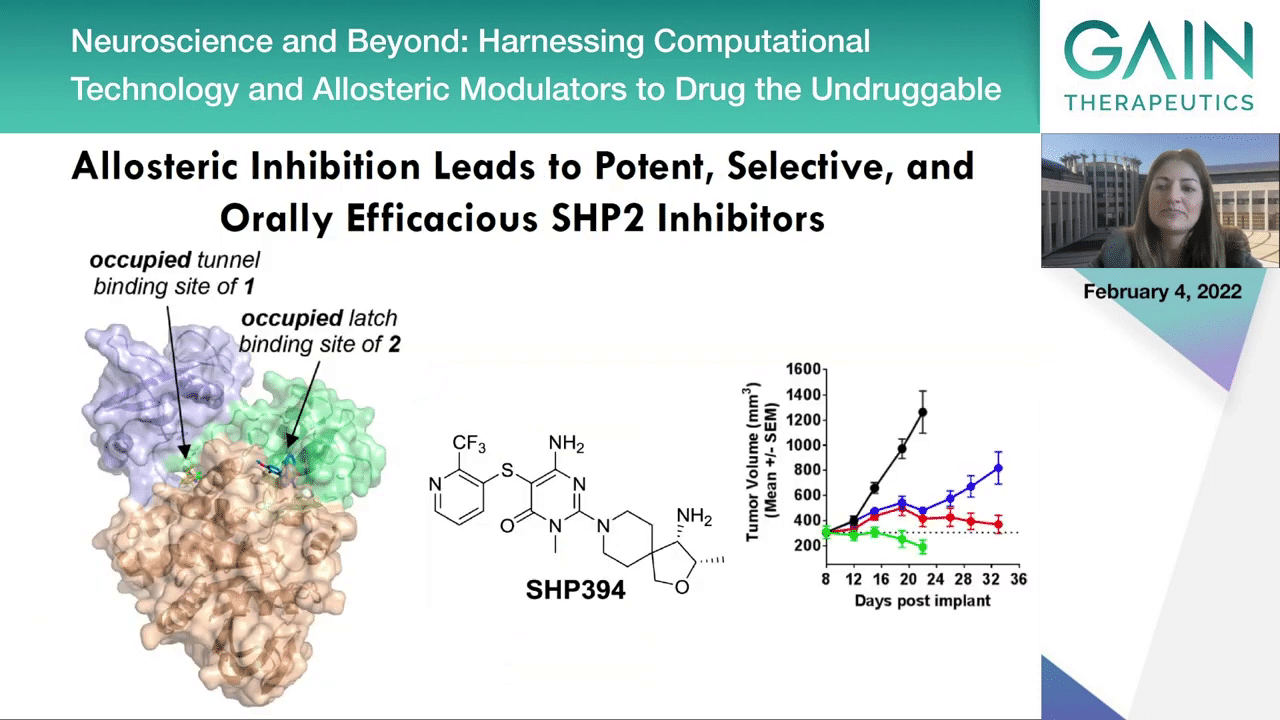
First, the protein tyrosine phosphatase SHP2 is an oncoprotein and also a immune modulator because of its role in the program cell death PD-L1/PD-1 pathway. This study published by Novartis in 2019 in the Journal of Medicinal Chemistry showed that this phosphatase SHP2 can be targeted not only by one but actually with two allosteric inhibitors in two different distinct sites and distinct from the active site, and showing in this approach that with allostery it is actually possible to develop compounds with good pharmacokinetics and cell activity, which has been a major challenge for phosphatases to this date.
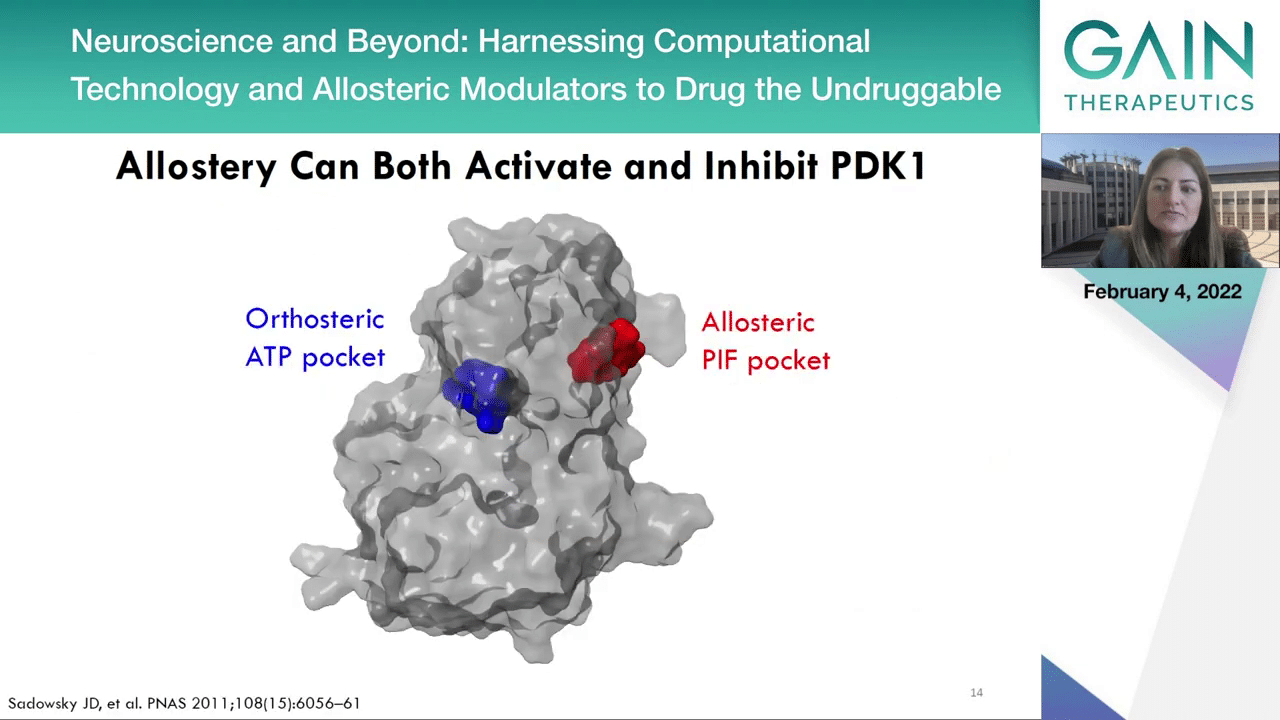
In the next slide, we can see another exciting case study which is the modulation of PDK1, which apart from its active site it also has a distant regulatory site termed PDK1 interacting fragment or PIF pocket.
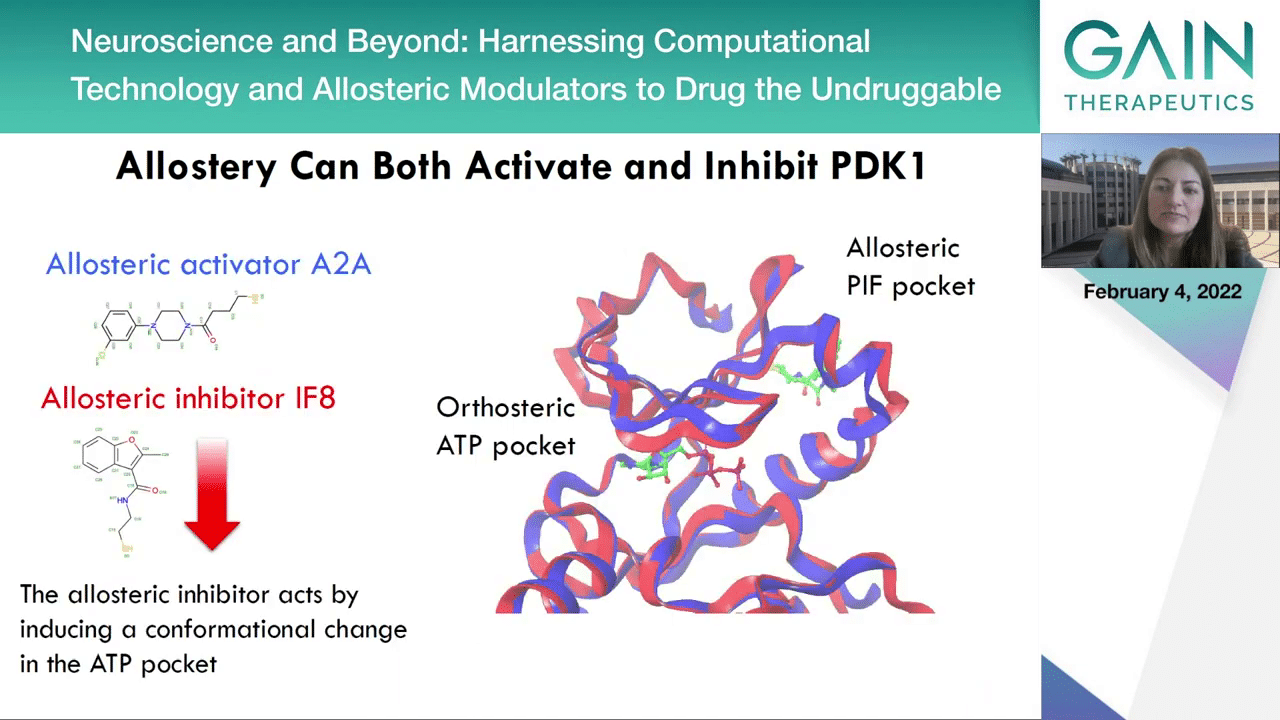
In the next slide, we can see that in fact using the same, the very same allosteric pocket but a different ligand in its pocket every time, one can achieve either activation of the protein with this positive allosteric modulator A2A that you can see on the left, or an inhibitor, an allosteric inhibitor which you can see in red in the IF8 molecule. The activator enhances the binding of the substrates in the orthosteric pocket, facilitating their reactions and thus activating the enzymes. The inhibitor, the allosteric inhibitor which binds at the same pocket acts by inducing a confirmational change in the ATP pocket blocking the catalysis. PDK1 is a great example for illustrating because activating a kinase has not been possible in any other way except allostery.
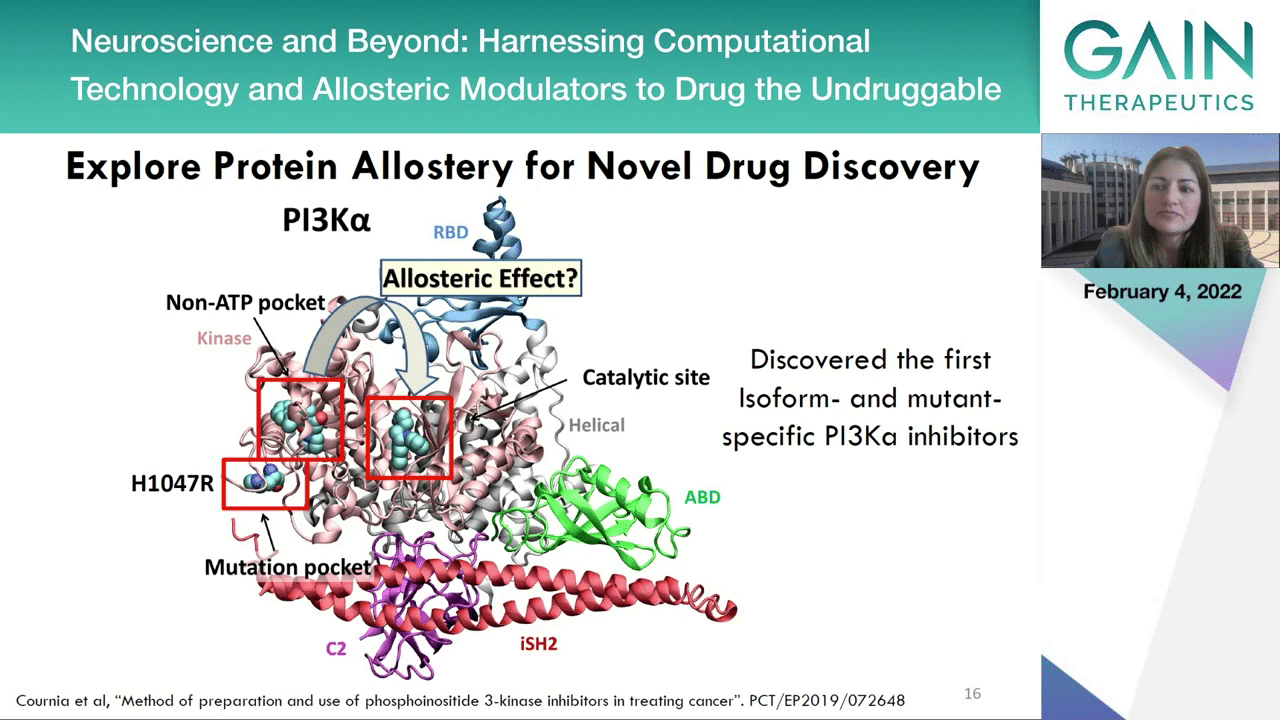
In the next slide, we can see one of our own examples on modulating the activity of PI3K alpha, a known oncogene, which a first isoform and mutant-specific PI3K inhibitor discovered, showing excellent in vivo efficacy against PI3K mutant-dependent tumors in genetically modified mouse models, and allosteric site is more specific to this individual protein, potentially leading to safer medicines because as it is isoform-selective, it cannot inhibit PI3K beta, gamma or delta.
The next slide. How does the phenomenon work exactly?
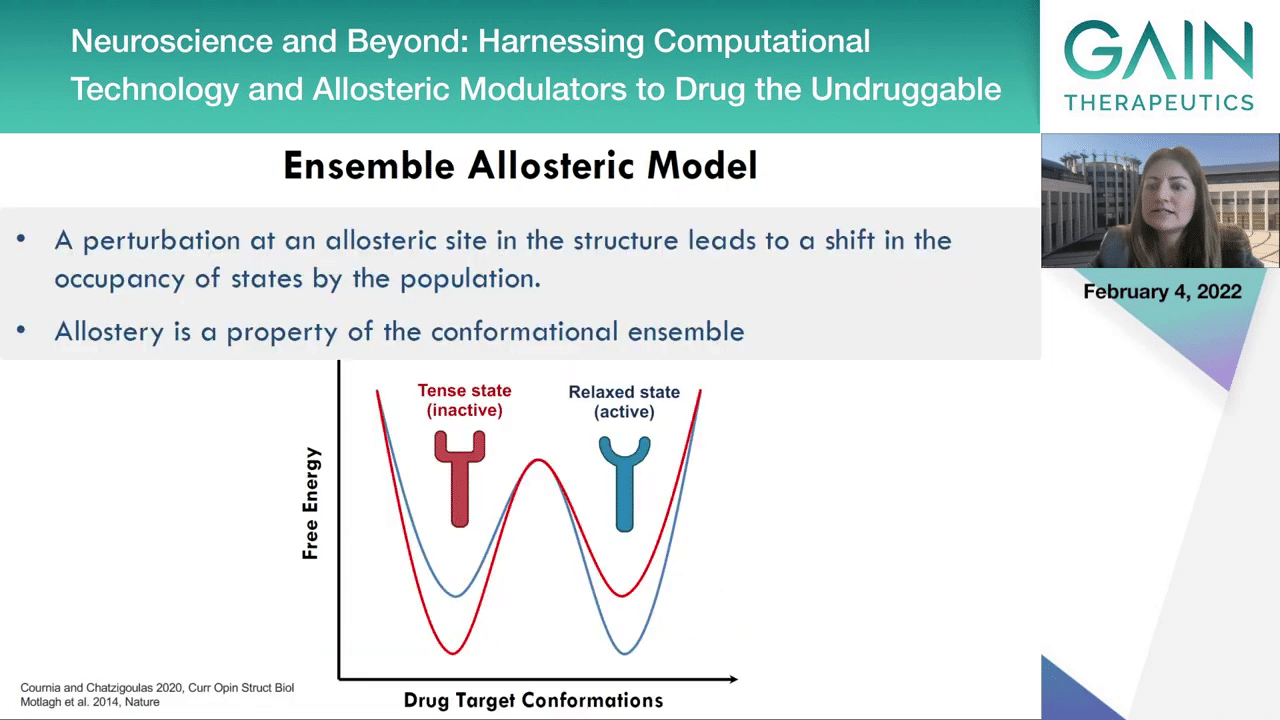
Actually, the active and the inactive states of the enzymes exist in the population of the proteins. It’s something that we call the confirmational ensemble.
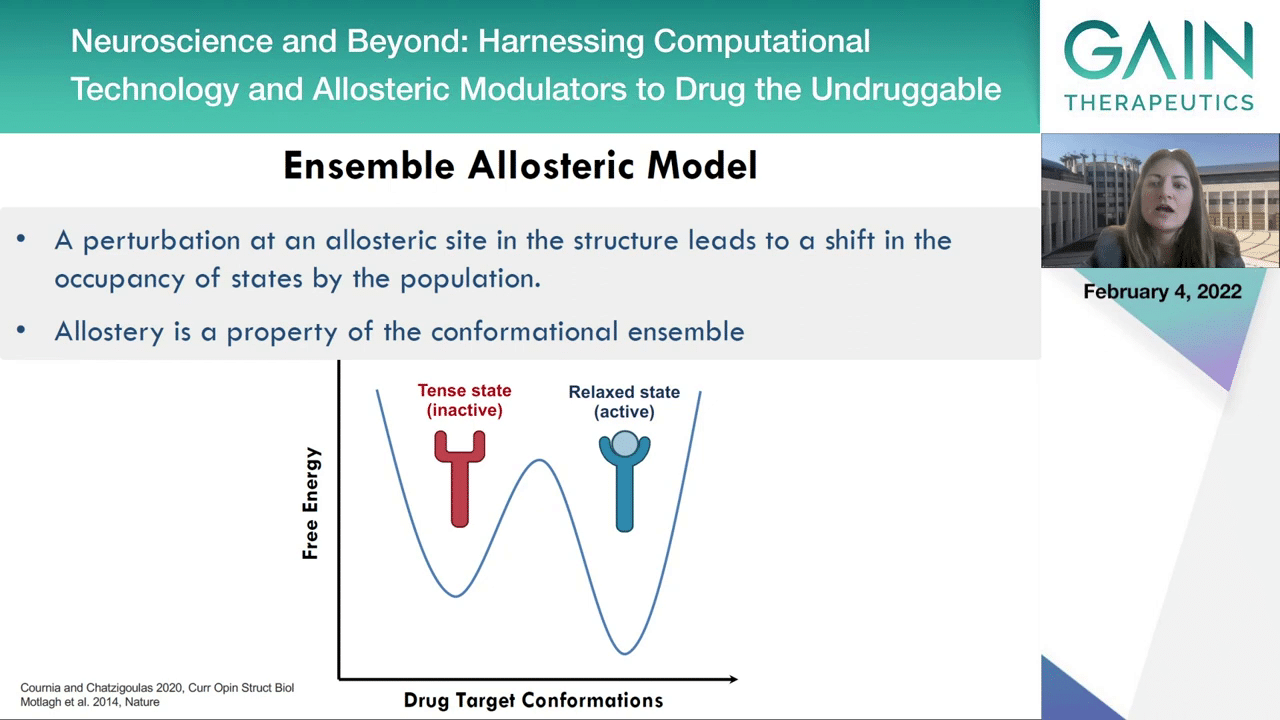
On the next slide, an allosteric activator can stabilize the active state and an allosteric inhibitor can then stabilize the inactive state.
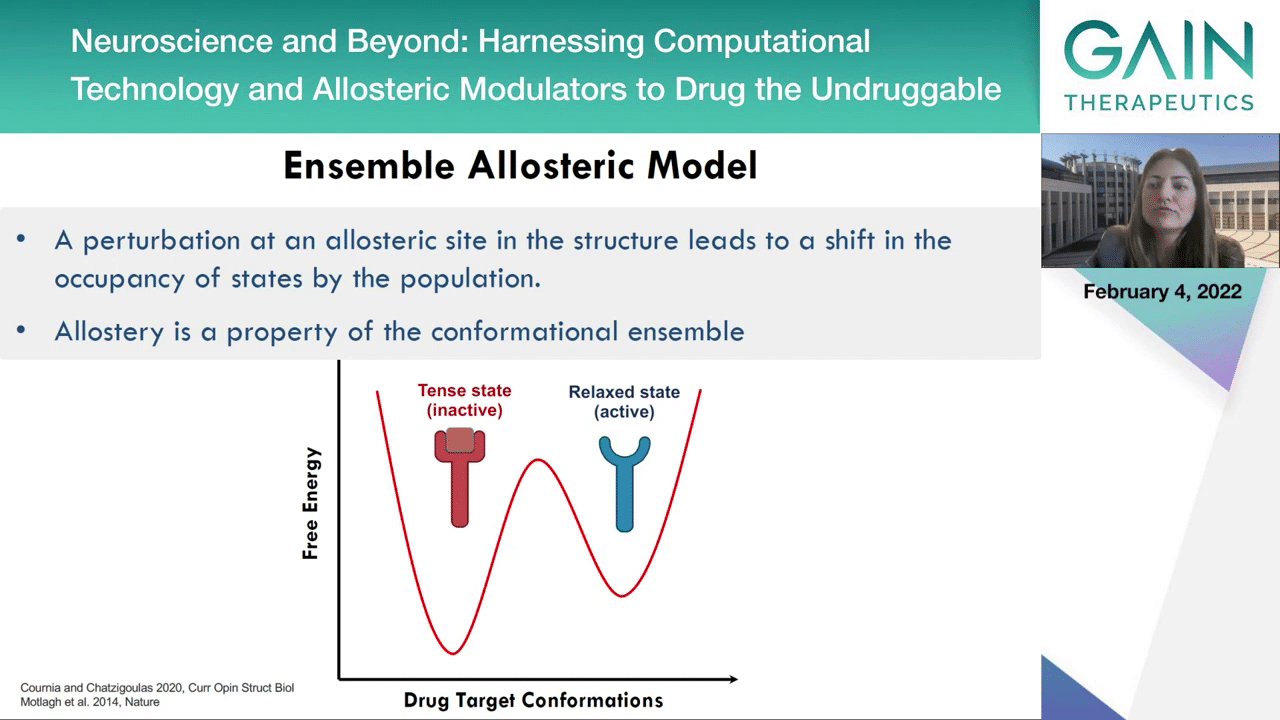
Therefore, allostery is the property of the confirmational ensemble and it can be assumed there may be a property of full dynamic properties.
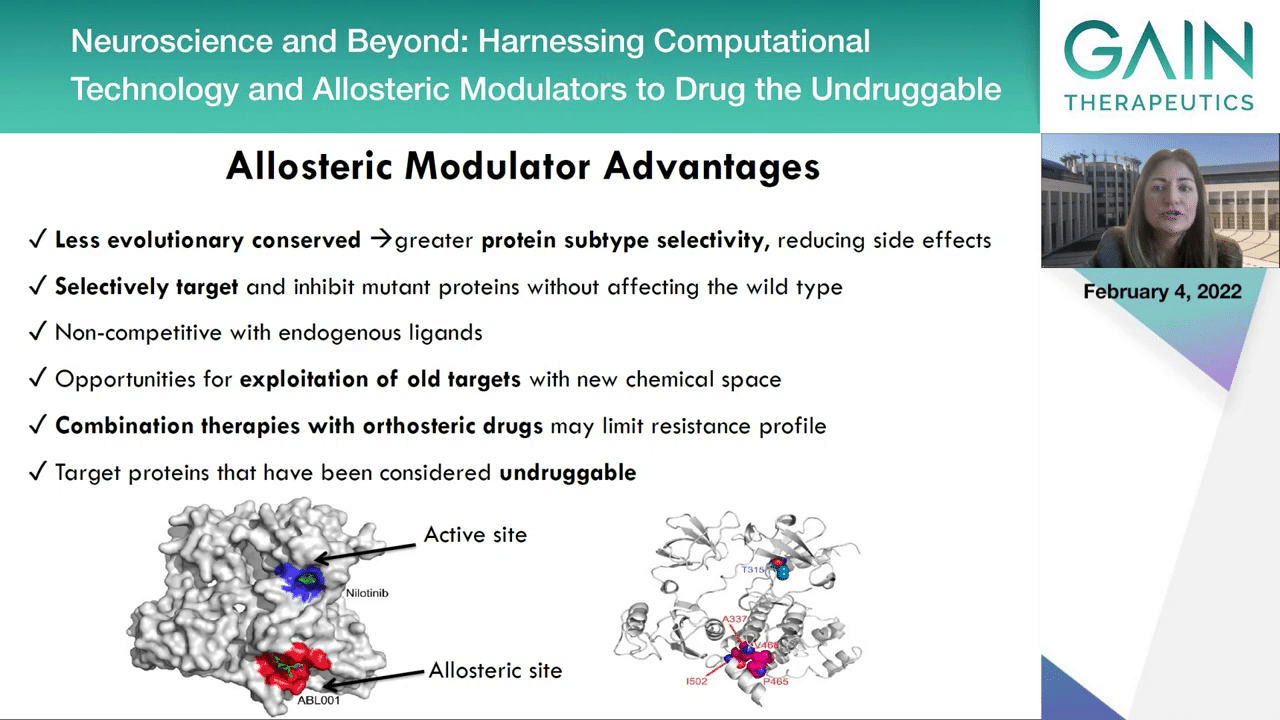
In the next slide, we can see that there are numerous advantages to designing and applying allosteric modulators.
First of all, the allostery binding sites are less evolutionary conserved which can lead to greater protein subtype selectivity, reducing side effects. We can selectively target and inhibit mutant proteins without affecting the wild type, as it’s not going to target the orthosteric sites. Allosteric inhibitors are non-competitive with endogenous ligands which may lead up to 1,000 of increase of the in vivo efficacy. We have opportunities for exploitation of old targets with new chemical space. Combination therapies with orthosteric drugs may limit their resistance profile, and actually, we can target proteins that have been considered undruggable because now we can discover new binding pockets as the second secret of life.
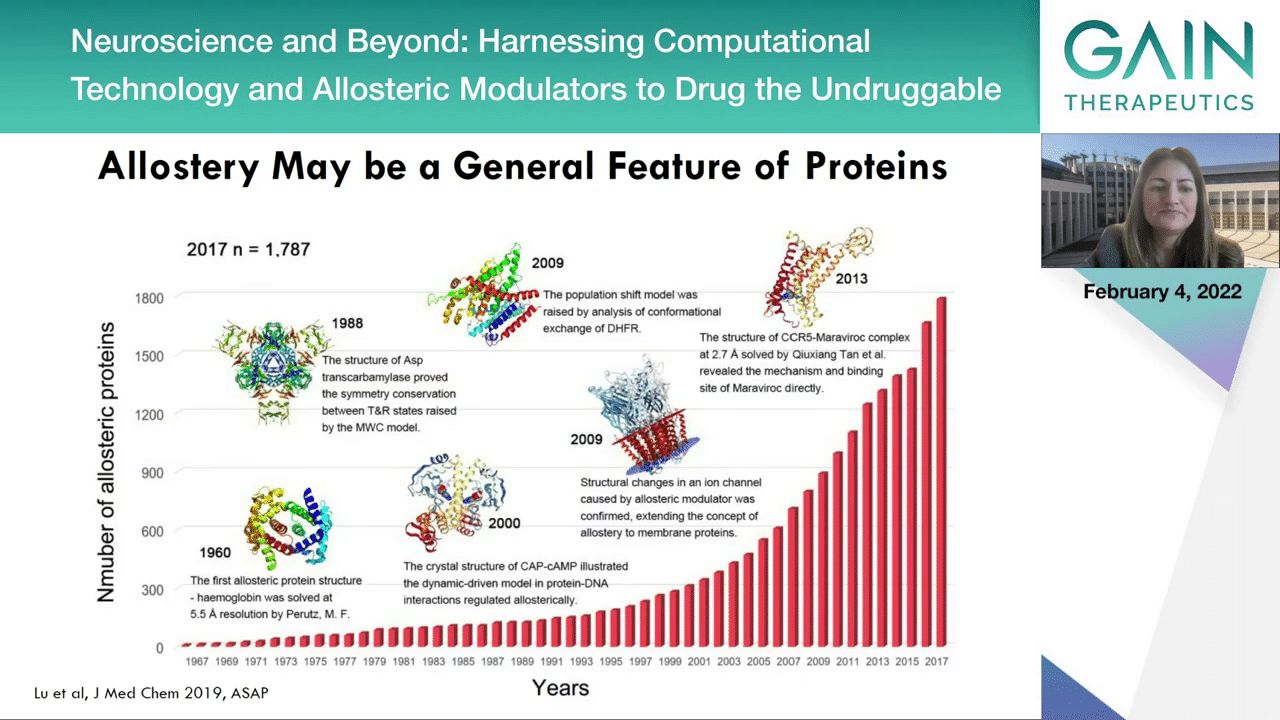
As I mentioned, allostery may be a general future of proteins and as we didn’t know of this phenomenon, at least not until 120 years ago, we can now see that the literature is really expanding exponentially in this field. We don’t know it yet but allostery may well be a general future of the proteins.
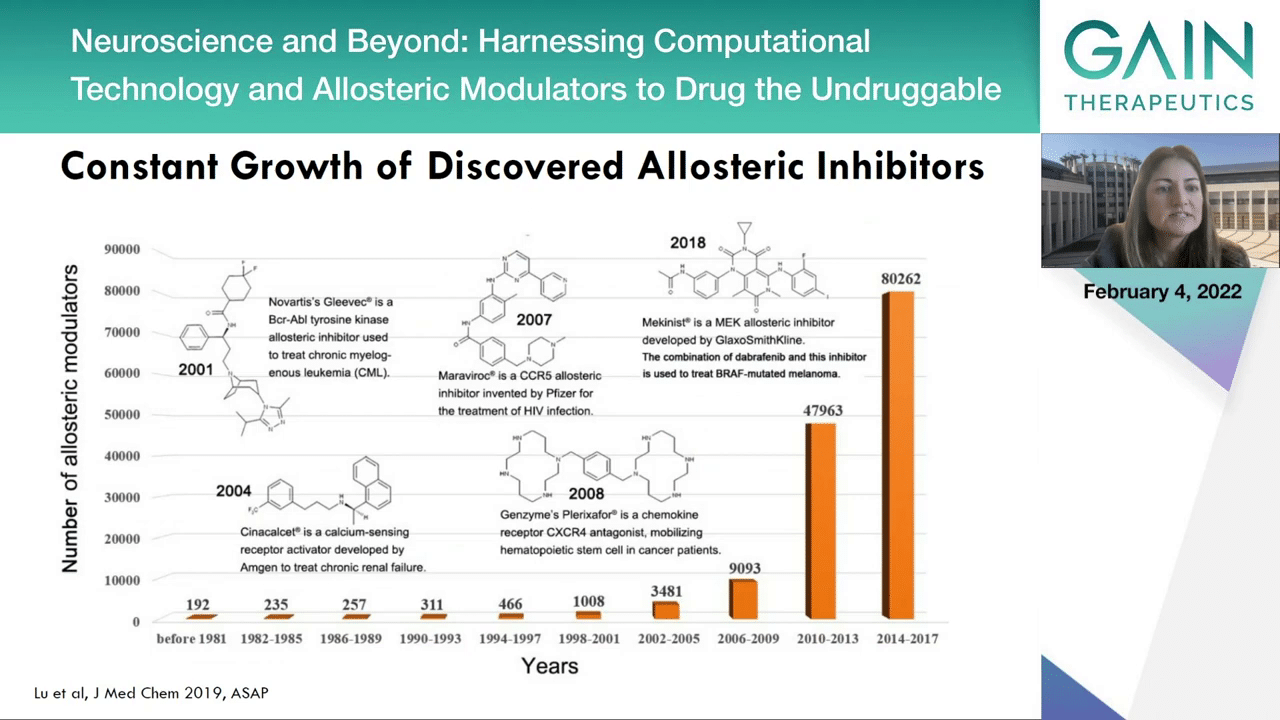
The next slide we can see that also there’s a constant growth in the literature of discovered allosteric inhibitors.
The next slide? I think there’s one slide before that that has a movie. After that I think there is a movie that should play. I don’t know if the movie is playing because I can’t see it.
Moderator
Dr. Cournia, the movie is playing on the playback screen.
Zoe Cournia
Oh, the movie is playing. Okay, great.
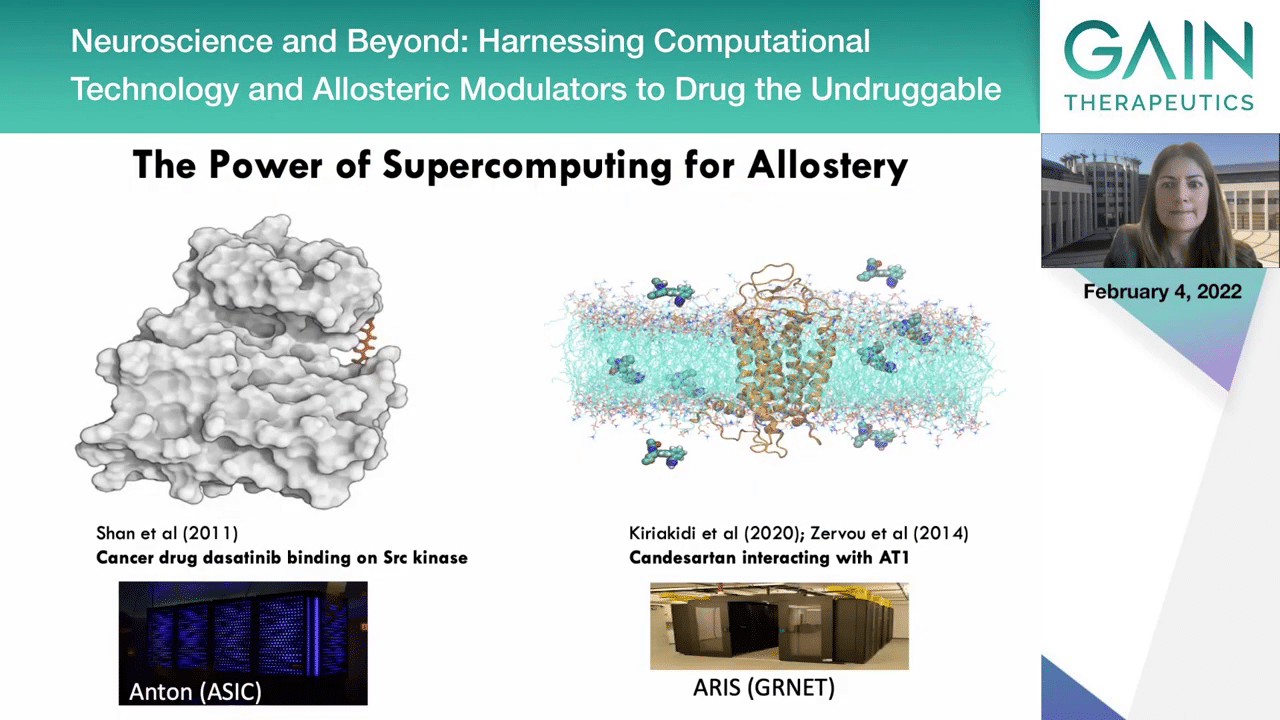
If you could play both movies, we can see in this slide that combining the power of supercomputing structural data, several publications have shown that the binding sites of proteins can be discovered, as you see in these two movies of molecular dynamic simulations where the different ligands explore the surface of a protein to discover their binding pocket. Actually, these are brute force simulations that depend on supercomputing power and they don’t harness any other intelligence, computational techniques that we may have and that were mentioned in the beginning.
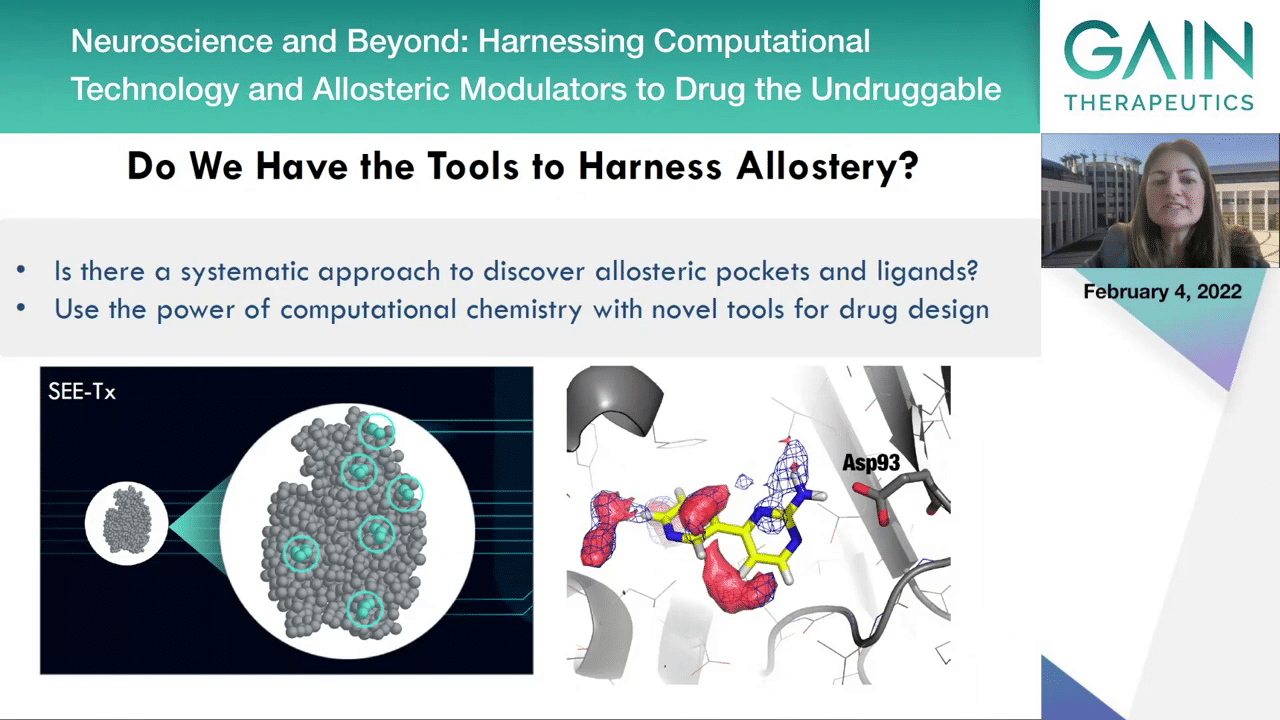
When we go to the next slide, we can see that aside from supercomputing, can we actually have the tools to harness the allostery and is there a systematic approach to discover allosteric pockets and ligands? In fact, the answer is yes. With the power of computational chemistry with novel tools for drug design, for example using the SEE-Tx methodology to discover new allosteric binding sites and predicted druggability, we can see that with this tool we can discover—you see them in light blue, new binding sites. And actually, on the picture on the right, you can see that with mixed solvent molecular dynamic simulations you can see that the organic solvents as our sample, they actually map very well the potential molecules, the potential new candidate drugs that can bind to this new pocket that are revealed through this mixed solvent molecular dynamic simulations.
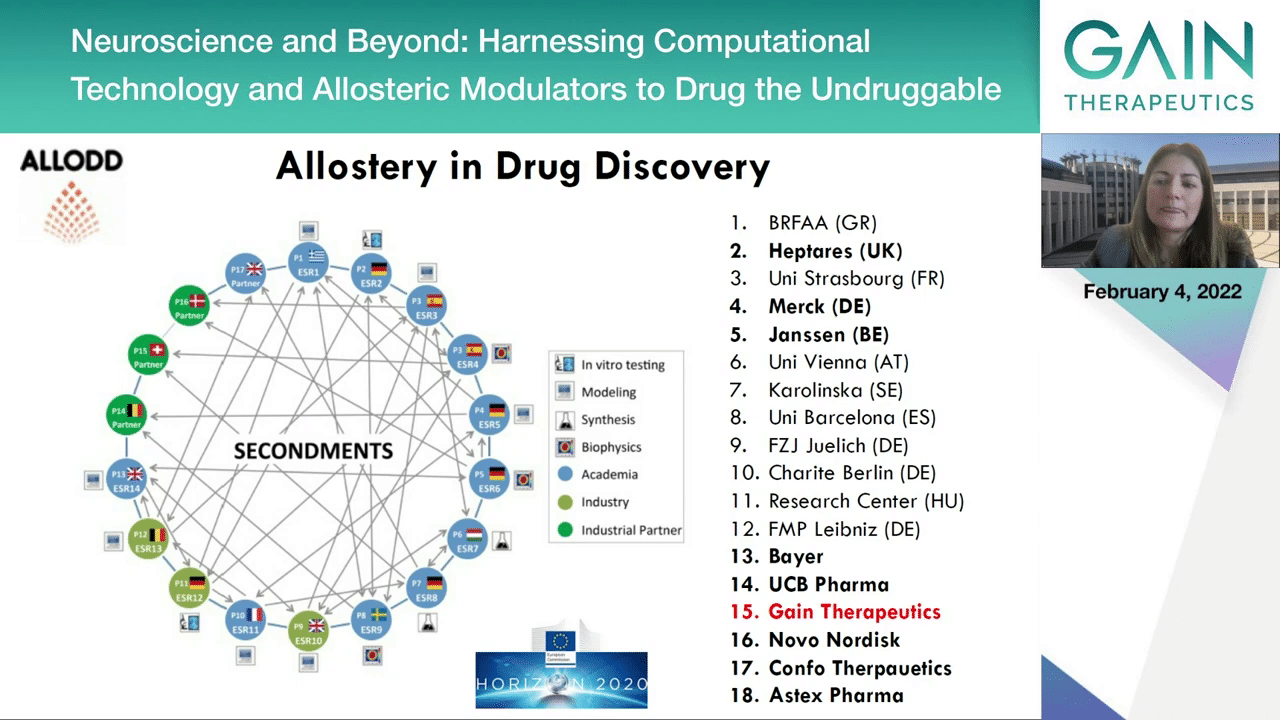
On the next slide, in fact this wonderful technology was one of the driving forces to work together with Gain Therapeutics in a European Commission funded program with €3.6 million combining different pharmaceutical companies, as you see here on the right, as well as academic institutions to apply this technology and other technologies and train the next generation of scientists in allostery and drug discovery.

It is my strong belief that the landscape of new classes of drug therapies will come from allosteric drugs. While drug discovery is challenging and has encountered many successes, of course, but many highly costly failures, the allosteric drug space has barely been explored. The fact that many proteins are considered undruggable does not imply that this is indeed the case. Transient allosteric pockets, such as the ones revealed with the technology I just showed you, in this undruggable proteins or in proteins with which the druggable proteins associate, directly or indirectly, can and will lead to therapeutic advances. Without doubt, allostery will lead the drug discovery efforts of the future.

The next question is—next slide—do you want to be part of this future? Thank you very much.