
Transcript of the presentation is as follows :

We’re a publicly traded company, so I will be making forward-looking statements today. There are risks about these forward-looking statements that we have described in detail in our SEC filings. So please read up on those and keep them in mind as you make investment decisions.

A Gain Therapeutics is essentially focused around three areas of expertise and competence.
One is the computational drug discovery platform that we have established, that our chief technology officer has developed over a period of 10 years and that we have applied to generate the entirety of our product pipeline. The platform is particularly suited to identify novel targets binding sites on proteins of interest that are involved in the disease and to identify small molecule binders that then interact with these binding sites and it’s particularly suited to identify allosteric binding sites and I’ll talk about that as well a little later in the presentation and then the third leg that were standing on is our robust product pipeline.
We have two lead programs one in Parkinson’s disease that I will talk about in this presentation but also one in Gaucher disease that’s related. It’s called here about that in a few minutes and both of them are moving towards the clinic or the upcoming period. We have Global Partnerships. We have a strong network of academic collaborators but also a partnership with saint Alice Pharmaceuticals on the drug discovery program in oncology where we are applying our platform to identify targets, binding sites, and small molecules. Mm together with Saint Alice strong management team. The company is quite young was established in 2017, but all of us on the management team have been in other places before and so, the accumulated experience is actually quite impressive. And we bring that to Bear for the company. And as mentioned, we have the lead programs moving into the clinic in the upcoming period.

In terms of value generation opportunities, there are really two probs that were focusing on:
One is our discovery platform our See-TX Discovery platform. It’s one that we have used, as I mentioned to generate our existing pipeline, we continue to apply it to generate additional pipeline programs for ourselves. While, we’re also using that pipeline in collaborations, with Pharmaceutical companies. I just mentioned Saint Alice before that drug discovery tool. Drug discovery collaboration with these Pharma companies, in its heavy effort, very heavy focus of our business development efforts as well.

The other area of value generation is our programs, the pipeline and here, bit more detail a bit larger for all of you to see, I did mention the Parkinson’s program. It’s a, it’s a focused on genetically caused this form of Parkinson’s disease. It’s there relevant genus GBA1 and a mutation of the GBA1and the which is the origin of Parkinson’s symptoms in these in this patient group, a similar mutation is also applicable for our Gaucher disease program, which is how these programs are very closely related to also focused on a GBA1 gene mutation.
We have additional programs in lysosomal storage disorders, a program in metabolic diseases, specifically liver and Lung and then we have Discovery efforts in oncology as you can see, from the for the breadth and range of these programs. It is all based on our platform that really has applications across all therapeutic areas.
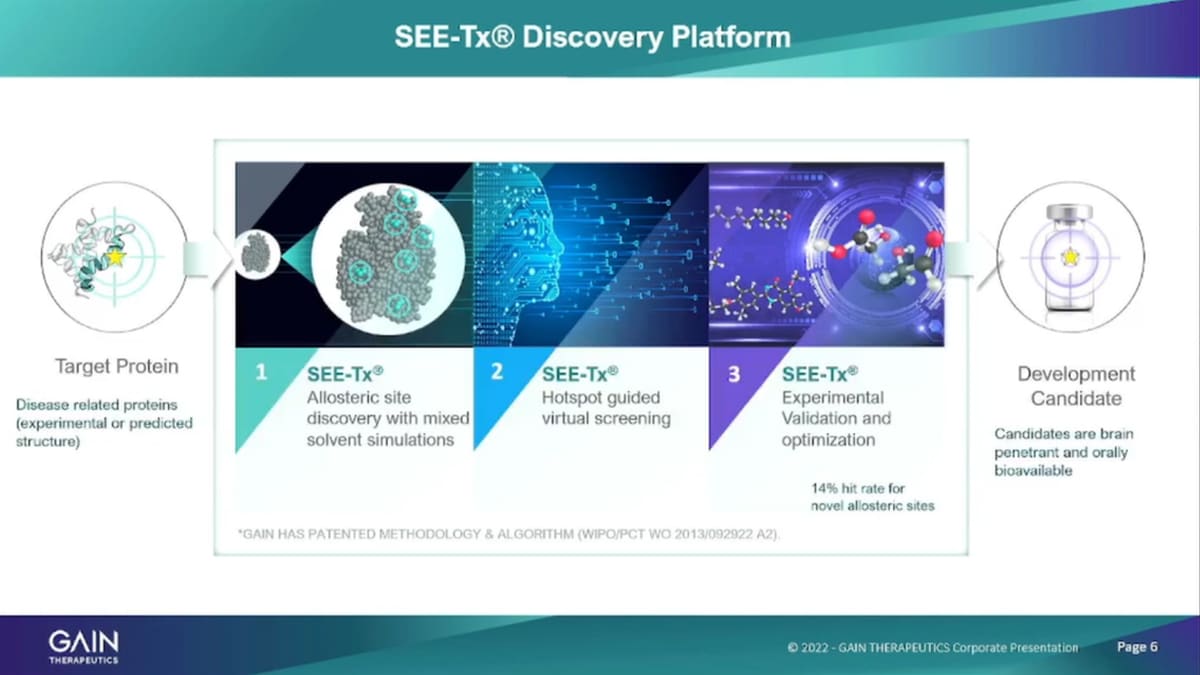
The platform is physics-based and structure-based platform. So, it’s technically not artificial Intelligence, where you have to rely on a lot of existing information to be absorbed and then processed, we’re using a physics-based approach. So, we’re looking at the protein itself and the structure and identify binding sites with our algorithms on those on that on those protein. So, what we start out with is the target protein and we just need the structure of the protein that can either be an experimentally derived structure through whatever experimental methods are available to identify protein structures. We also can use predicted structures. So Alpha fold for example, is that open-source database of protein structure predictions that is accessible to everybody really has opened up the entire universe of human proteins that are now available to us for use in our platform and our drug discovery efforts. The first step in the platform in the drug discovery applying SEE-Tx drug discovery is as the, it’s a binding site identification. So we are running the models the algorithms to identify with Organic solvents and number range of organic solvents to identify binding hotspots.
We analyze those binding hotspots, at an atomic level, to identify the binding sites that are most suitable for pharmaceutical intervention. And then, we apply the platform to identify to do a virtual screening program about 10 million compounds and that’s in step two, here to identify binders to small molecule binders to those hotspots and binding sites that we have identified. We take about Out we’re use about 10 million compounds to run that screen of those 10 million. We typically take about 100 compounds then into experimental validation, which is a very important step because all the predictions that you can make with a computer model, ultimately are only valid if you can actually confirm them in real life experiments and that’s what we’re doing in our labs in Barcelona. So, we take about 100 molecules per screening run on a particular protein to the experimental validation confirm the binding confirm the effect of the small molecule on the protein and then take that into further traditional development process.

The platform is highly efficient and effective so it takes us only one to two weeks, to identify key to getting from a protein structure to having identified binding sites. These are binding sites that have never been seen before but these are all novel binding sites and takes one to two weeks to come up with, suitable, binding sites on a protein, we then run the virtual screening process that takes three may be four weeks and then the experimental validation assume the assess are all up and running takes two to three weeks. And so, the output of the platform is about 14% is a hit rate of about 14% more talking about hit rate. This is not so, the virtual it right, it is 14% of the hundred molecules that were testing experimentally are actual binders, and have an actual effect on the protein and then serve, as the basis for further development, characterization need series creation and so forth.
Maybe, some chemistry. That compares, If you’re looking at traditional methods high-throughput screening takes, maybe 18 months to two years to set it up to run the screening, then to remove all the false positives that are generated through high, put through screening campaign, then you end up with a hit rate of 0.1 percent, we do the same thing in, you know, less than three months. And have a hundred fold increase in terms of the output that we get from the platform.

A bit more specifically there’s a lot of people who are in computational drug discovery. We have several areas where we are differentiated from other companies for using molecular dynamics mix simulations, where were using a range of organic solvents to probe the protein surface to identify where these solvents interact with the protein service surface that panel allows us to It’s already do a density map of interactions. We then go, through a process of analysing these interactions, and make quantitative predictions at up, to the down, to the atomic level about what binds where?
On a particular protein surface, and it allows us to really identify the binding sites that are most suitable for pharmaceutical intervention. And then, in the virtual screening, the phase we’re going through the typical sort of docking analysis or looking at how molecules bind. But in addition, we’re also looking at how molecules let go. So, the undocking protocols are absolutely critical for us to identity to screen out false positives because you need to know how well the product binds a compound also how it lets go and the combination of information, gives you the best sense for what could be a suitable drug candidate.
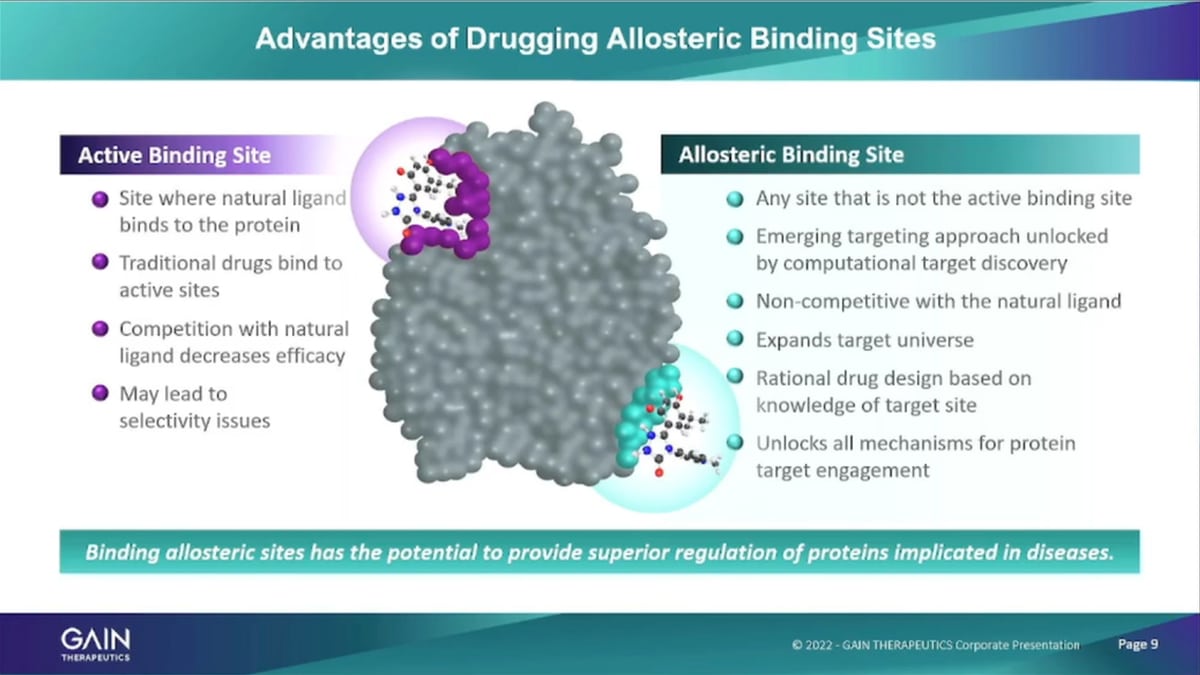
The second area mentioned that were using the platform a just because described is the identification of allosteric binding sites on proteins of Interest. Allosteric binding sites are essentially is any place on the surface of a protein. That, is not the active binding site. And the active binding site is the place where the natural ligand of a protein binds, if traditional Pharmaceuticals. They typically, go to the active binding site because that’s the known site that can be actually explored for pharmaceutical intervention through traditional methods. And then you have the prob, the issue that the potential issue that you need to overcome, which is the competitive binding. So, the small molecule that the Pharma company wants to develop as to out-compete the Natural ligands to engage with the natural binding sites and that needs to lead to selectivity issues, potency issues and so forth. Focusing on allosteric, binding sites that don’t have any natural binders in the body that we don’t. We avoid that that issue entirely.
Also, as I mentioned, we can target any protein of interest at this point about 90% of proteins. Do not have a known a binding site. And with our platform technology with SEE T-x we’re scanning the entire surface of the protein and are able to identify binding sites on proteins. That don’t otherwise have anything any place else that is known that it’s that is suitable for binding. Umm In addition, looking at allosteric binding, Mm it opens up a whole range of ways of interacting with the protein. The traditional Approach of binding to the active binding site. Essentially have either an activation of the protein, or looking to inhibit the protein activity. So, it’s activation or in Innovation. And these are the two ways that traditional Pharmaceuticals interact with proteins.

Through an allosteric binding molecule, we have been many additional ways to interact with the protein. So for example, we can stabilize a misfolded. Protein and restore its function, which is the approach that we have applied to all of our lysosomal storage disorder. Programs also applies to our Parkinson’s program. We can also then look at destabilizing doing the opposite, destabilizing the protein.
So as we’re looking at molecules that bind the protein, it’s a molecular Dynamic simulations of the protein moves about, we can find molecules that bind into cryptic pockets of the protein that are otherwise, not see able. Stabilize the protein in a misformed way. That then causes the protein to be either no longer functional or two to be degraded in the cell. We can also go for specific targeted degradation through protac approach and then the more traditional inhibition and activation approaches by binding in close proximity to the active binding site.
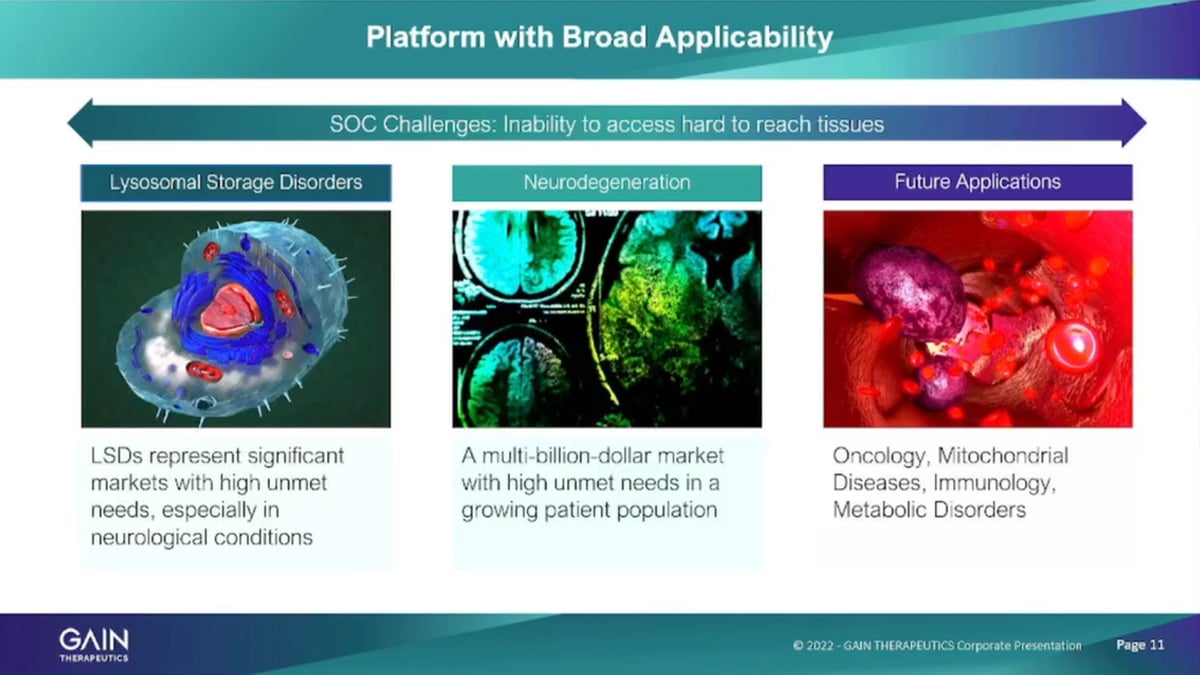
The company as I mentioned has focused initially, on lysosomal storage disorders. It’s a great area for small company to work in. These are all the rare diseases, very high unmet medical need and the current therapies such as in enzyme replacement therapies that help these patients are typically not able to or are actually not able to cross the blood-brain barrier and using a small molecule approach that we have with our binders we actually have the ability to cross the blood-brain barrier and also address neuropathic Symptoms of lysosomal storage disorders that are currently untreatable. The program in Parkinson’s disease is based on a related mechanism.
So that’s the newer degenerative program that we have it’s based on a related mechanism of lysosomes of the Gaucher programs or the lysosomal storage disorder program. But, it is obviously a different patient population. I’ll talk about that in just a minute. And then, we have other applications of the platform that we’re exploring actively now in oncology in metabolic diseases. But, also across the full spectrum of the therapeutic areas because the platform is entirely agnostic as to therapeutic areas structure based physics, based approach, which was need to know what the protein structure is, and we can run the program and identify a binders against these proteins.

Switching gears and focusing on now primarily on our Parkinson’s program, which is the most advanced in our pipeline Parkinson’s, is a disease, that affects about 10 million people worldwide, about 14% of those have a specific mutation of the GBA1 Gene, and as a result of that mutation, Developed Parkinson’s symptoms, the these types of patients the GBA. One Parkinson’s patients have an earlier onset of the disease and the much faster progression of the disease than idiopathic Parkinson’s patients.
And so as a result of a much increased need for for a pharmaceutical therapy that actually can help stop the disease progression. Gaucher disease, is that they’re related program. It’s also based on the mutation of the GBA1 Gene, and is a rare disease childhood disease. And add one that program is in close, close follower in terms of moving into the clinic.

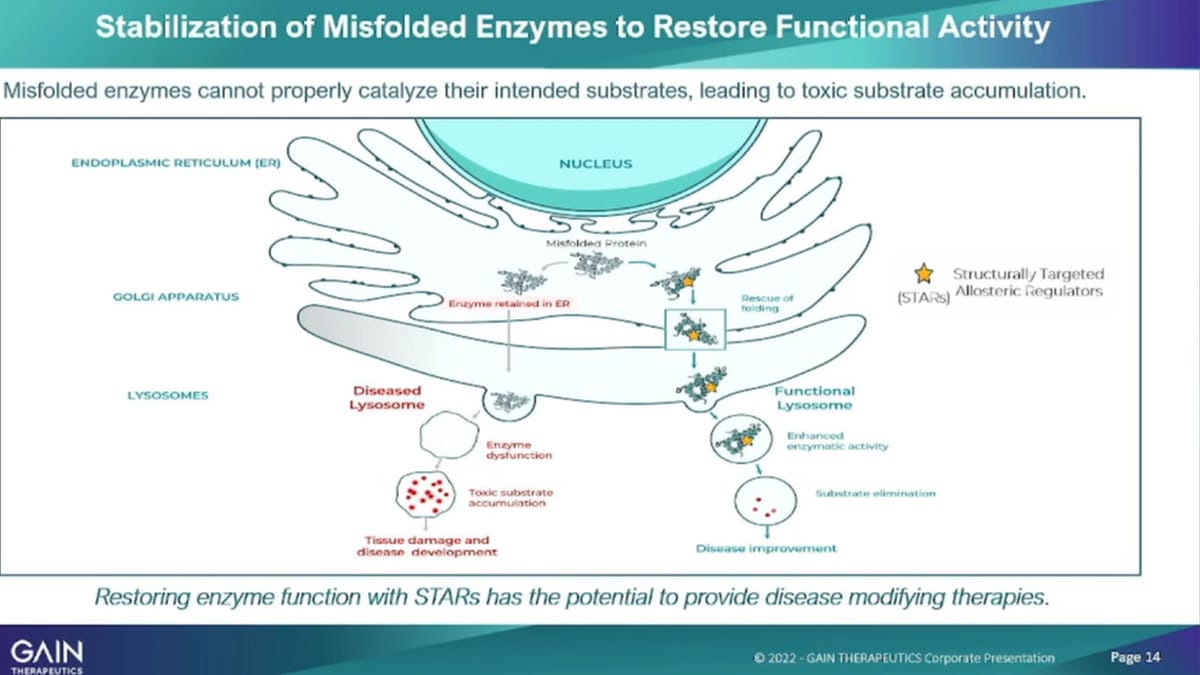
For both programs, just giving a bit of a survey as a science refresher on how the mechanism of what an enzyme does and how our mechanism of action of our molecule actually works. So, in a normal cell, you have a GPA1 gene that’s not mutated, that is functioning. Well it expresses an enzyme called gk’s and gk’s, the job of G case is 2. Then once it’s created in the endoplasmic, reticulum move into the lysosome. Lysosome is the wastebasket of a cell and then the in the lysosome that enzymes job is to remove toxic materials. So, you can imagine if you have a misfolded enzyme that is not able to. As a result of a GBA1, mutation you have a misfolded enzyme that enzyme then is not able to move into the lysosome and is not able to perform its job there, as a result. You see an Accumulation of toxic materials in the lysosome, which then ultimately causes cell death. And if that cell death happens in a dopaminergic, neuron will cell as a result in the brain. You see every reduced levels of dopamine and ultimately the development of Parkinson’s disease, What we’re doing with our structurally targeted? Allosteric Regulators.
Our lead compound for the Parkinson’s program is were binding to that misfolded enzyme. So we’ve had that, we have used a platform to identify an allosteric binding site. You’ve used the platform to identify the small molecule, that we’re now developing our that small molecule binds to that to that enzyme to the misfolded enzyme. In real life, stabilizes the enzyme enables that enzyme to actually move from the endoplasmic, reticulum into the lysosome. And, there perform the function that we had, that it’s required to do, which is to reduce the toxic materials. We have shown in vitro, and in Vivo, all three elements of that postulated mechanism of action, which is enhancement of enzyme levels, after administration of our compound.
In the endoplasmic, reticulum We then see enhanced levels of enzyme in the lysosome and we then see reduction of the toxic materials in these cells. And, then in Vivo, we see increased cell, survival increase, survival of dopaminergic neurons and increased dopamine levels as well as a reduction of inflammatory markers that reduction of inflammation in the brain, which is another indicator that what we’re doing actually, The wars since lysosomal, he’ll sell health and enhances cells survival and as such that the therapy that we are developing has the potential to actually halt disease progression. So, it’s not asymptomatic treatment or in acting at the very beginning of what causes the disease upstream and as a result create healthy cells that survive better.

We’ve tested this in in collaboration with Dr. Ricardo Feldman at the University of Maryland, he has developed models neuronal model. Spaced on patient-derived IPSC’s looking at dopaminergic neurons cortical neurons and other cell types and these tested our molecules in that model in his Labs at the University and I’ll show you some of the results.
In just a minute this is probably as close as you can get to a patient in a cell based model. So, people refer to this approach as a patient in a dish. And, so the results that we have generated in these models are highly relevant for the properties and qualities of the molecule that were developing.

So in these models, what we have shown is in going from left up and down to, right? We’ve shown increased GCase enzyme levels. We’ve shown increased trafficking’s of movement of the enzyme from the endoplasmic, reticulum into the lysosome. We’ve shown that depletion of the toxic substrates we’ve shown that depletion of alpha-synuclein, which is a Hallmark of Parkinson’s disease. And there’s company number of companies that develop products, specific solely on trying to remove alpha-synuclein for doing that with a much more natural process by just restoring enzyme function in the cell. We’ve shown overall improved enzyme function through increased alta fetchick flux in dopaminergic neurons, and we’ve also shown that reduction of inflammatory markers.
As I’ve mentioned, all of that shows that just going at the very beginning of the origin of where the disease starts and restoring enzyme function restores lysosome and he’ll cell health and has all the beneficial effects of having a healthy cell, which is you don’t have inflammation, the cell survives longer and does that work that you do as including for dopaminergic neurons that creation, the manufacturing of dopamine for the brain.
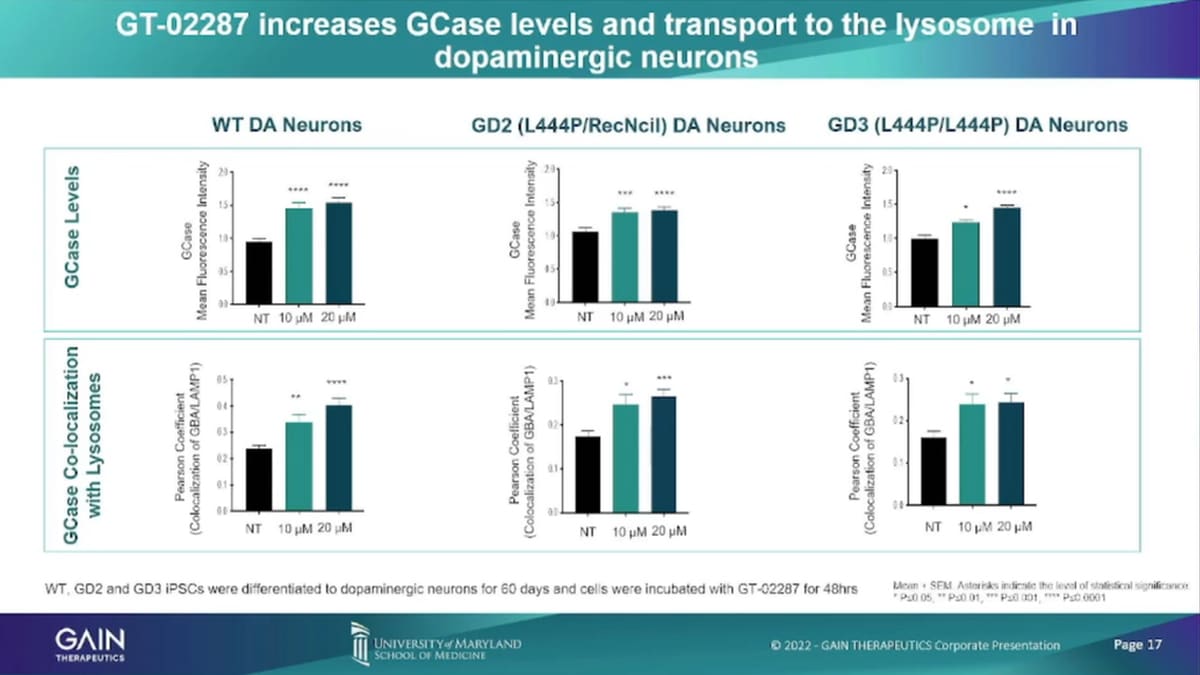
So here’s some of the specific data and I won’t go into too much detail on those. So if we’ve seen here, as statistically significant levels of increase of GCase levels, localization of GCase in the India enzyme.

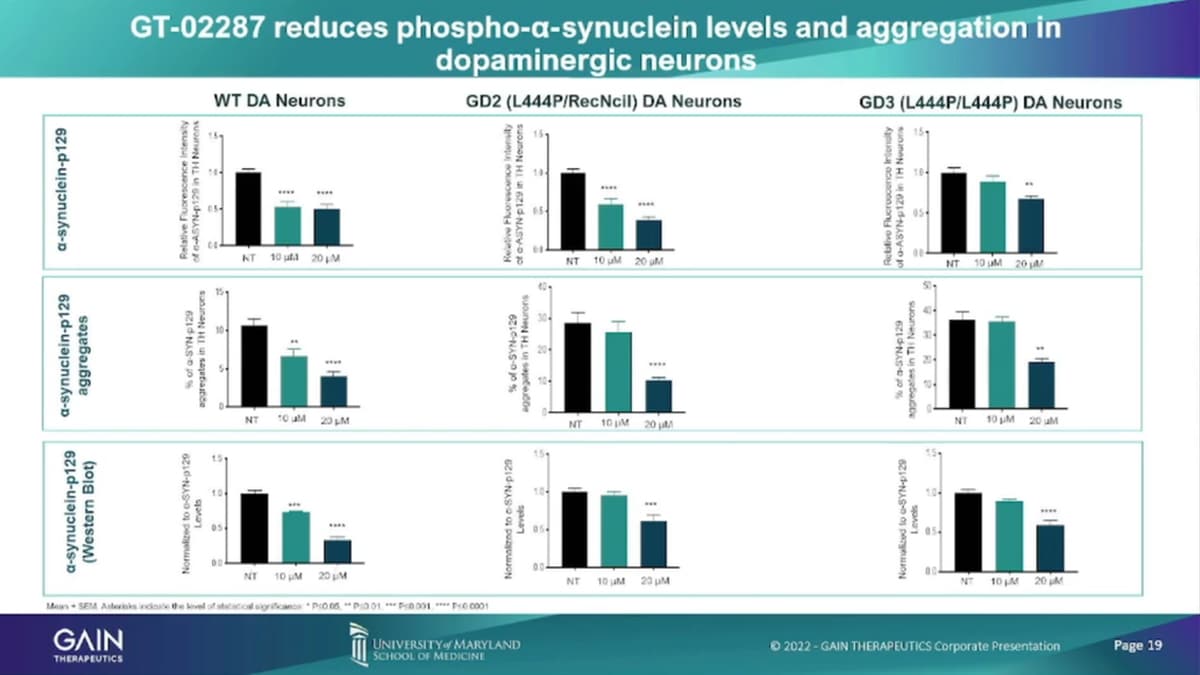
Similar than in cortical neurons. We’ve seen the reduction of the accumulation of the toxic substrate Glucer, and as well as the reduction of alpha-synuclein, both the phosphorylated in aggregate accounts.

And here in conclusion of the cell based data, we’ve seen the very comparable effects and the results of a reduction. Also, in the inflammatory markers still restoring cell here as an indicator of restoring cell health.

We also have run a number of animal models. I’m going to just show you the Rotenone Rat model, Rotenone is that’s a proof of concepts in Vivo studies that we’ve done. Rotenone is essentially, it’s a herbicide that if administered to an animal causes Parkinson’s-like effects and symptoms. And, so the test then is you have controlled healthy animal. You have series of animals that have been affected with the Rotenone and are sick and then you treat the number of these rats, with our molecule.
And, what we have shown in these animals is very consistent with what we shown in the cells in that, we are depleting, the toxic materials, are very improving neuronal activity. And then, now in animals, we can also observe the Locomotion Parkinson’s being a movement disorder and we see improvement in Locomotion in these animals, as well.

Hear, a bit of the that the data on that specifically, on alpha-synuclein, as I mentioned that key biomarker for the development of Parkinson’s disease. And, we have we seen as the tips statistically significant reduction in animals in these Trojan on injured animals, through the administration of our molecule.
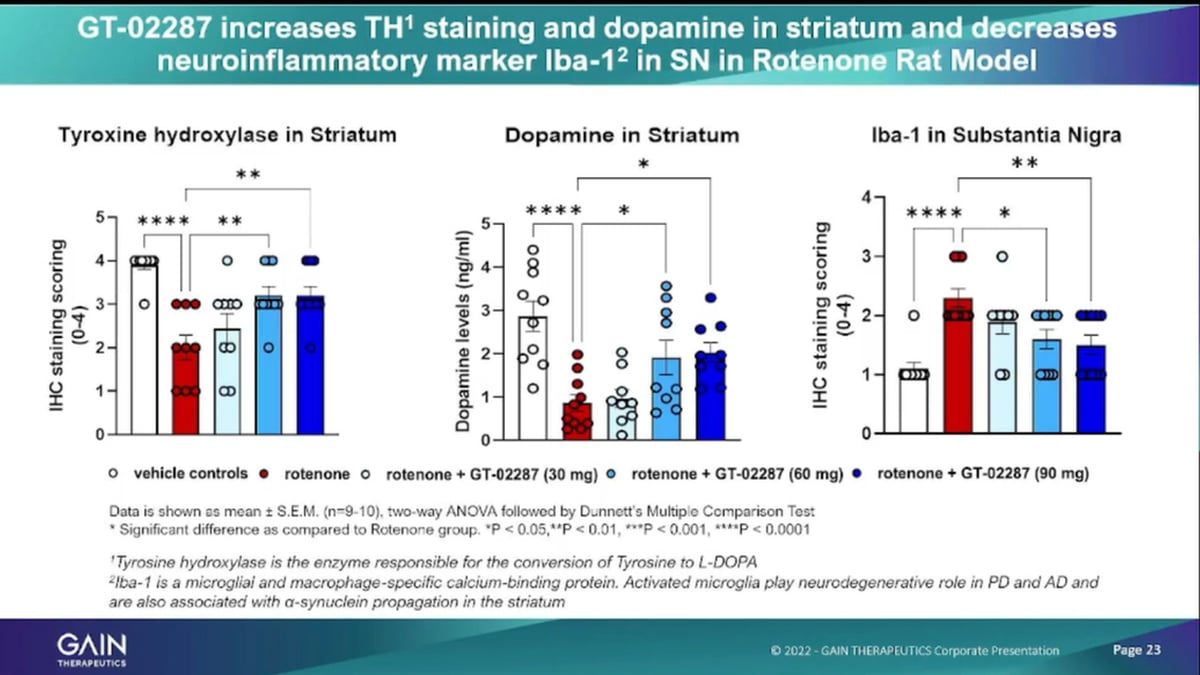
Similar then for the inflammatory markers, the very right side, on being such a marker. But more importantly, to me, is the tyrosine. Hydroxylase staining that’s an indicator of dopaminergic neurons cell survival and then the related effect of increased dopamine levels. And you see on both fronts administering, our lead compounds to Injured rats, roten on injured rats as actually statistically significant effect of improving the survival of dopaminergic. Neurons and increasing at the dopamine production.

And here’s the, let’s see now unfortunately, my tech has left now, are you still there? Sorry. Can you run the video here and you need to click on all three to make it to make it work. So on the left, you have the vehicle control meaning the healthy rats in the middle You have the rat injured with the Rotenone. And on the right on the right, you have the Rotenone injured that has been treated with our compound mass As you can see, the vehicle that control and the treated rat behave, very comparably in terms of their movement in this box.
Whereas, the Rotenone injured rat that has not been benefited from a treatment is actually very slow, and if you’re looking at the tracing map on the right there you can see. So, very comparable traces on top for the vehicle and at the bottom for the treated rat, whereas, the middle right in the middle, the injured rat that has now been treated has a lot less movement including the walking around, as well as the rearing and getting up on hind legs which is for us, the first real evidence in Vivo of the activity. The desired activity of ours, these compounds in a proof-of-concept animal model.

So based on these very encouraging data were actually very happy to move forward with that program, the Parkinson’s program and take that into the clinic. In the upcoming period, we continue to focus on our platform. Look to exploit that platform both internally for creating our own programs but also to generate value through Partnerships focusing on Allostery, which is a novel relatively recent Focus. Area of pharmaceutical development, mainly because previously was not you weren’t able to identify allosteric binding sites in a very directed way.
We can do that with our platform and then we’re looking at exploiting our platform, our programs, our pipeline, both by Progressive them ourselves, but looking at partnering opportunities for those programs. With that, I’m happy to conclude. Thank you, for your attention and you have any questions happy to take them after?
