
Introduction:
Below is a transcript of a conversation Professor Roy Alcalay at the KOL event.
Operator: Good day and thank you for your patience. Welcome to today’s video webcast event Gain Therapeutics KOL Analyst Event: Beyond Symptomatic Treatment of Parkinson’s Disease. Today, you may submit online questions at any time using the window on the webcast. Please note that today’s event is being recorded.
Before we begin, I would like to read a brief statement. Some of the statements during the course of today’s event will be forward-looking statements. These forward-looking statements are based on the company’s expectations and assumptions as of the date of this event. Each of these forward-looking statements involves risks and uncertainties that could cause the company’s future results to differ materially from those expressed or implied by these forward-looking statements. So the company is providing here the customary Safe Harbor disclaimer regarding these forward-looking statements.
At this time, I would like to turn today’s conference over to Hartaj Singh of Oppenheimer & Company. Please go ahead, sir.
Hartaj Singh: Great. Thank you, Chris. Thanks for making this easy. These things are never easy, actually. So thanks for making this as painless as possible. I’m one of the biotech analysts at Oppenheimer & Company. My name is Hartaj Singh, and our team covers Gain Therapeutics. Today, we are actually fortunate to have three great speakers ahead of us. We have presentations from Professor Roy Alcalay, Professor Tiago Fleming Outeiro and Dr. Joanne Taylor, who’s at Gain Therapeutics. They will go in that order.
Dr. Roy Alcalay: Thank you, and thank you for inviting me to speak today. So I was tasked with a relatively simple task of a talk and that is to talk about the GBA-associated Parkinson’s disease and other clinical neuropathies, current unmet medical needs and future therapies.

Key Insights from Dr Roy Alcalay
Genetic Basis for Focusing on GBA
Dr. Roy Alcalay: Thank you, and thank you for inviting me to speak today. So I was tasked with a relatively simple task of a talk and that is to talk about the GBA-associated Parkinson’s disease and other clinical neuropathies, current unmet medical needs and future therapies.
Exploring GBA as a Promising Target
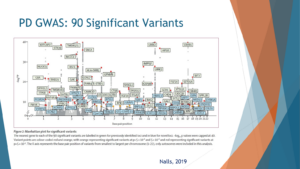
Now why look at GBA, there are 90 hits on the GWAS, on the Genome Wide Association Study for Parkinson’s. This is taken from NOSM colleagues from 2019.

So when we look at the genes for Parkinson’s in this figure, we understand why GBA and LRRK2 pop up. In the x-axis, we’re looking at how frequent are the mutations in the general population. And in the y-axis, if you have the mutation, what’s your risk of Parkinson’s? The vast majority of genetic risk factors for Parkinson’s aggregate in two places; those in the bottom right, they’re coloured green here, are variants that are relatively common, so 20% to 30% of the population can have them. They increase the risk by 20% or 40%. So instead of 1% of people with PD, 1.2% of those are carriers. So very common, but the effect size of a mutation or a variant is not that high.
At the top left, there are variants or mutations, if you have them, your risk for Parkinson’s is very, very high. Like people who are homozygous carriers for Parkin, almost all of them will develop Parkinson’s. But these mutations are exceedingly rare. LRRK2 and GBA are located somewhere in the middle where they’re relatively common, common enough to start thinking about clinical trials, and their risk enough, they have enough of an effect size that it makes sense to modify this effect to reduce Parkinson’s risk or to intervene on it. And that is why I think GBA as a gene is a great drug target.
GBA Mutation Prevalence and Phenotype

So my brief talk will be focused on why focus on GBA, and I’m basically going to talk about five points. First is the pathogenic variance in GBA is the most common genetic alteration identified in Parkinson’s disease. The second is that GBA, or GBA1 and its new name, is very similar to [indiscernible] Parkinson’s disease. So clinically, it looks the same, which holds promise that treatments for GBA may later be helpful for people with GBA. And when we look at them under the microscope, that’s the third point, the diseases look very much alike, the genetic and non-genetic. The fourth point that makes GBA an interesting target is that we know what happens when people have two mutations in GBA in the disease that is called Gaucher. A lot of research from the Gaucher world is research we can use now for understanding GBA PD. And that there have been already clinical trials for GBA PD, so they’ve demonstrated feasibility already. So it’s not like, oh, this is not doable. Not only that it’s doable, people have done it already.
GBA Precision Medicine: Feasibility and Future Prospects

Starting with the fact that there are the most common genetic alterations in Parkinson’s, I will show some information from the PD Generation Study. The Parkinson’s Foundation funds the PD GENEration Study and the goal is to make genetic testing and counseling accessible for people with PD and for their doctors. The idea is that if people know this genetic data, people if they don’t know it, and they won’t know how to act on it. And specifically for the talk today, if people don’t know that they’re carriers, they won’t participate in the study. We realized that once we offer people genetic testing and counselling to explain to them the genetic results, a lot of people are interested in getting and obtaining the genetic results, and they basically line up to participate in the study.
GBA Mutation: Clinical Implications
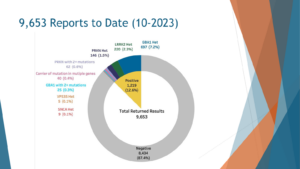
So the cut data of October 2023, less than four years of study, more than roughly 10,000 people have been genotyped and received their results back. Out of those nearly 10,000, 10% or 12.6% received a positive result, the vast majority of those who received the positive result received it in the GBA1 gene. So 7.2% of all comers and this is unbiased recruitment, we then encouraged anyone to participate, 7.2% of people with Parkinson’s disease harbor a GBA mutation that would make them eligible for a GBA2 intervention. In this case, all of them received the results back and I assume that some of them are just waiting in line for a clinical trial to start.
Pathological Insights into GBA Parkinson’s

So the second point, now that I showed you that it’s relatively common, we have at least one study from PD GENE, 700 people who know their genotype and they were interested in knowing their genotype, so they may act on it, acting by means of participating in studies, let me tell you a little bit about the phenotype of people with GBA1 Parkinson’s. So just a word back, as I said, roughly 5% to 10% of people with Parkinson’s carry a GBA1 mutation. In selected populations, specifically Ashkenazi Jews, the risk is much higher, so over 20% of people of Ashkenazi Jewish ancestry with Parkinson’s carry a GBA mutation. But carrying a GBA mutation doesn’t mean Parkinson’s, it means that it increases the risk of Parkinson’s. So the penetrance is estimated at roughly 10% to 30%, it depends on the studies. Probably closer to 10, if you ask me.
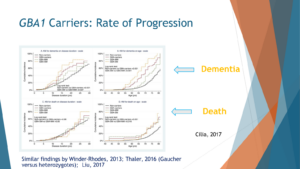
Once people develop Parkinson’s, their Parkinson’s progresses at a faster rate. So this is a paper from Celia and colleagues. It’s an Italian study that looked at outcomes that are considered bad outcomes: dementia, death, and other people showed with motor outcomes. We see that people with GBA mutations progress faster, which means they reach those bad outcomes of dementia or death faster than people with Parkinson’s without these mutations. They also showed a dose effect. We’ll talk about it a little bit later when they have a severe GBA mutation and one that is probably bad for you, worse for you, the time to reach dementia or death and others showed it with motor performance happens faster the more GBA burden you have. So idiopathic PD progresses slower, mild mutation faster, severe mutations even faster than that, and two mutations the fastest.
Understanding GBA Pathophysiology

When we look at it as a clinician, I’m also a clinician, on an individual level I cannot take a look at someone and say you have a GBA mutation. But if we look at people as a group, they have Parkinson’s which looks like Parkinson’s progresses a little bit faster. Parkinson’s nowadays is not just defined by the motor symptoms that people think about when they think about Parkinson’s, like tremors and slowness. There are actually non-motor symptoms, a battery of non-motor symptoms associated with Parkinson’s, including cognitive impairment, hypoxia, reduced sense of smell, acting out in dreams which is REM sleep behavior disorder, and autonomic dysfunction that would be clinically presented with constipation, lightheadedness when changing positions. All these symptoms are common in idiopathic PD, but actually even more common in people with the GBA mutations in Parkinson’s.
Future Therapeutic Avenues
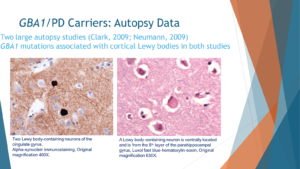
When we want to look at the ultimate phenotype, not just how the person experiences Parkinson’s but also how the clinician perceives their Parkinson’s, when you look under the microscope, what do you see, we see the Parkinson’s in GBA PD is very similar to idiopathic Parkinson’s. So I’m showing you two slices from the brain. One of them on the right, which is in pink, is the typical staining of the brain which is called hematoxylin and eosin. And in this one, as you see, just in the middle you see a Lewy body. A Lewy body is an accumulation of proteins, specifically Alpha-synuclein in the cells, that’s typical for idiopathic Parkinson’s disease, but this brain is taken from someone with GBA in Parkinson’s. When we stain with antibodies, specifically against Alpha-synuclein, and that’s the staining to the left, we see that these Lewy bodies in the person with GBA PD are indeed positive for Alpha-synuclein.
In two studies, one from England and one from Columbia University in New York, when the autopsies of people with GBA PD were compared to idiopathic Parkinson’s without GBA PD, those with GBA mutations had more burden of this protein of Alpha-synuclein and more Lewy bodies in their brains.
GBA is relatively common among idiopathic PD patients. Clinically it looks the same, progresses faster. Progresses faster is bad news for the patients. Good news for people who design clinical trials because outcomes can be reached faster. But it’s not just clinically, also under the microscope, the diseases look alike.
Insights from Gaucher Disease
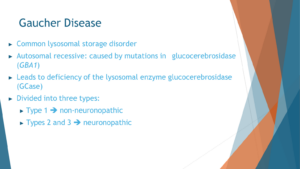
The next point I’d like to emphasize is the significance of the GBA gene, which is not obscure but rather well-known in the medical community. GBA has garnered attention primarily because inheriting two mutations in this gene leads to the development of Gaucher disease, a prevalent lysosomal storage disorder. Alongside Fabry disease, Gaucher disease stands as one of the most common lysosomal storage disorders.
To understand Gaucher disease, it’s crucial to grasp the role of the lysosome, often referred to as the cell’s recycling bin. Within this cellular compartment, proteins and lipids marked for breakdown and recycling are processed. In Gaucher disease, the enzyme glucocerebrosidase (GBA), responsible for breaking down specific lipids, is defective due to mutations in the GBA gene. Consequently, lipid accumulation occurs.
Gaucher disease is categorized into three types. Type 1, the most prevalent form and notably more common among Ashkenazi Jews, typically allows individuals to live into old age. This is because lipid accumulation primarily affects peripheral organs such as the liver, spleen, and bone marrow, sparing the brain. The specific lipids that GBA should break down are known in this context.
However, when Gaucher disease affects the brain in addition to other areas, it is termed neuronopathic, leading to type 2 and type 3 variants. Type 2 presents in infancy and type 3 manifests in older children. In these cases, neurological symptoms become pronounced and severe, including seizures, myoclonus, cognitive decline, and ataxia, alongside the peripheral manifestations observed in type 1.
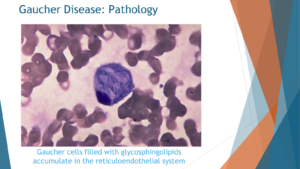
Upon microscopic examination of individuals with Gaucher disease, a distinct observation emerges: the presence of macrophages laden with accumulated lipids. Specifically, these lipids accumulate within the macrophages at the center of the cellular composition. This visual insight provides a clear understanding of the consequences when the GBA enzyme, also known as glucocerebrosidase enzyme, fails to function properly. In instances where this enzyme is completely deficient, lipids known as glucosylceramide and glucosylsphingosine accumulate within the affected cells.
Fortunately, Gaucher disease does have available treatments. However, a significant challenge arises in the context of Parkinson’s disease: the treatments administered for Gaucher disease are typically delivered peripherally, either intravenously or orally. Unfortunately, these treatments do not effectively cross the blood-brain barrier, thus failing to alleviate the burden of reduced glucocerebrosidase levels within the brain.
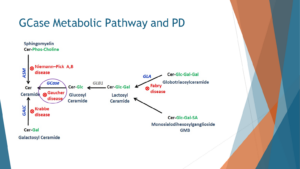
A cursory examination of the metabolic pathway associated with GBA sheds further light on the intricacies of the condition. This pathway, known as the ceramide metabolic pathway, unfolds within the lysosome. When the GBA enzyme (highlighted in purple) malfunctions, the lipids it is designed to degrade, including glucosylceramide and glucosylsphingosine, accumulate upstream within the pathway.
Additionally, highlighted in red are other lysosomal storage disorders, including Krabbe disease, Niemann-Pick disease, and Fabry disease. While these disorders are also linked to an increased risk of Parkinson’s disease, none rival the prevalence and impact of Gaucher disease.
Loss of Function Hypothesis vs. Gain of Function Perspective
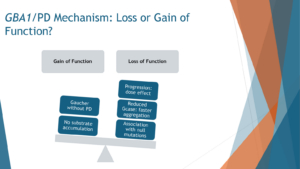
The prevailing notion regarding the connection between GBA mutations and Parkinson’s disease centers on the concept of loss of function. Essentially, when mutations occur in the GBA gene, resulting in impaired functionality of the encoded protein (the enzyme), the risk of developing Parkinson’s disease escalates. This loss of function hypothesis gains support from the fact that there are over 200 identified mutations in GBA, many of which have been linked to Parkinson’s. It appears that any mutation within the GBA gene elevates the risk of Parkinson’s disease, indicating a broader relationship between GBA dysfunction and Parkinson’s pathology.
Furthermore, the severity of the Parkinson’s phenotype correlates with the degree of enzyme activity reduction caused by the mutation. In other words, more severe mutations with diminished enzyme activity correspond to a higher risk and increased severity of Parkinson’s symptoms. Additionally, even mutations that lead to the production of non-functional glucocerebrosidase enzyme variants, known as novel mutations, are associated with Parkinson’s. This further supports the notion that reduced levels of functional glucocerebrosidase enzyme elevate the risk of Parkinson’s disease.
However, it’s essential to recognize that this loss of function theory is not exhaustive. There are alternative perspectives, such as the gain of function hypothesis, which suggest that there may be more to the relationship between GBA mutations and Parkinson’s than just enzyme activity. For instance, while individuals with Gaucher disease, who typically possess two GBA mutations, are at an increased risk of developing Parkinson’s, the majority of them still reach old age without experiencing Parkinson’s symptoms, despite having low enzyme activity. This discrepancy suggests that factors beyond enzymatic activity may contribute to Parkinson’s pathology in individuals with GBA mutations.
And then when we look under the microscope of the brain, as I showed you in the previous slide, we see the changes in the brain of people with GBA and Parkinson’s look like Parkinson’s, there’s Lewy body, with accumulations of protein. We don’t see the accumulation of lipids that one would have expected if the problem is that the enzyme is not working as well.

So a few more words about the glucocerebrosidase. It’s an enzyme that is reduced, that is completely diminished in Gaucher. People have shown in multiple, multiple studies, from competing groups in different continents, that reduced GCase activity, glucocerebrosidase activity, the enzyme that GBA encodes for, is present in autopsies of people with idiopathic PD. With GBA mutations that would make sense, but also people with idiopathic PD without the GBA mutations. In the blood, when we tried to measure it, the results are not as conclusive. But there is mild reduction, at least in a couple of studies, in glucocerebrosidase activity in idiopathic PD compared to controls. In people with GBA mutations, there’s clearly a reduction in glucocerebrosidase activity. And there’s some hints in the literature that the reduced activity is also associated with more severe motor phenotype.
Advancing Precision Medicine in GBA Parkinson’s: Targeting Pathways and Tailored Treatments
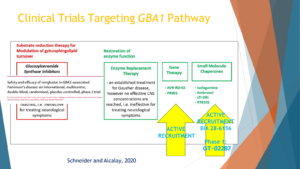
So the next point that I wanted to talk about, why focus on GBA1 and what’s missing, is the precision medicine studies in GBA1 have demonstrated feasibility. And I’m sorry, this slide the animation is a little bit broken, but the background slide is from a review that I wrote with Suzanne Schneider to talk about the different potential therapies for GBA. And I think the take home message from my topic of the talk that will convince you is that GBA is a great target. How to target it is open for questions.
One hypothesis, I’m going from the left to the right, option to how to treat the GBA pathway is instead of making the GBA enzyme work, the glucocerebrosidase enzyme work more, let’s make the counter enzyme work less, and that’s the glucosylceramide synthase. And there was a big Phase 2 clinical trial led by Sanofi that showed feasibility. They recruited hundreds of people with GBA mutations around the world, Europe, Israel, Japan, the North America, and the study didn’t show that the drug worked, but it showed that we can do precision medicine in GBA PD. It was just published in 2023 in Lancet Neurology.
And other ways to target the pathway would be to increase the enzymatic activity. That’s the second box, the first box in green. That works well in Gaucher. The problem with that for Parkinson’s is that those treatments currently don’t penetrate the blood-brain barrier, the enzyme replacement therapy that is used for Gaucher is too big to cross the blood-brain barrier. And, therefore, currently, that’s not an effective way.
The second box to the right would be gene therapy, like inject a gene with a healthy gene to the brain. There is an active recruitment for a study of the compound mentioned above.
And the last mechanism that I would like to mention is small molecules. These small molecules can be called chaperones, not all of them are chaperones, that can either take the GBA, the enzyme from the endoplasmic reticulum to the lysosome where it belongs in the recycling bin of the cell and make it work. Or, just one way or another make the enzyme work faster or better.
There’s one drug that was developed by LTI that is now in clinical trial in North America and Europe, working exactly in this mechanism, and the Gain Therapeutics component that will be mentioned later is also targeting this path here.
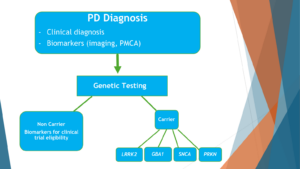
The idea is that currently people with Parkinson’s disease are diagnosed clinically and getting treatment by symptoms. I hope that this is dated and will change soon enough to a very different paradigm, where people with Parkinson’s disease will get a clinical diagnosis, will get a biomarker study, like a study of Alpha-synuclein that now works very, very well from spinal fluids, ideally from peripheral tissue in the near future, and imaging. Based on this, they will receive a Parkinson’s diagnosis. Based on the Parkinson’s diagnosis, they will get genetic testing. And based on the genetic testing, they will get the tailored treatment. And the first group that I would target is the GBA1 carriers because that’s the largest genetic group.
If a drug works with a genetic group, the next phase in that will be the last box to the left will be to look at non carriers of the gene and look at their biochemical portfolio, like their biomarker signature, to see whether they have biomarkers to suggest that one of the genetic drugs would work for them.
Conclusion

In conclusion, GBA mutations hold immense promise as a target for therapeutic interventions in Parkinson’s disease. Their prevalence in the Parkinson’s population, coupled with their clinical and pathological similarities to idiopathic Parkinson’s, make them an attractive focal point for drug development efforts. Insights gained from studying Gaucher disease, a condition caused by mutations in the same gene, provide valuable guidance for understanding the pathophysiology of GBA-associated Parkinson’s. Furthermore, recent advancements in precision medicine, including targeted therapies and gene editing techniques, offer new avenues for treating this subset of Parkinson’s patients. By leveraging these insights and technologies, researchers aim to develop tailored treatments that address the underlying molecular mechanisms of GBA-associated Parkinson’s, ultimately improving outcomes for affected individuals.