
Professor Tiago Outeiro who is Professor of Aggregopathies and Director of the Department of Experimental Neurodegeneration at the University Medical Center in Gottingen, Germany, and he will discuss the role of protein misfolding in neurodegenerative disease.
Watch the Full Video here:
Here is the Transcript of the Presentation by Professor Tiago Outeiro
I thank you so much for the introduction. As you heard, I’ll talk to you about different types of diseases, these are neurodegenerative diseases, and they are associated with the accumulation of misfolded and aggregated proteins in the brain.
As you heard, I am talking to you from Gottingen in the center of Germany, and we do our research here at the University Medical Center in the Department of Experimental Neurodegeneration.
We’ve focused on this topic represented here for many years now and the issue is to understand how proteins that are produced based on the information encoded in our genome become folded and functional, and we think it’s important to understand this process because we know that often there are problems along the way and this causes proteins not to behave and not to function properly, as we heard already in examples in other diseases before.
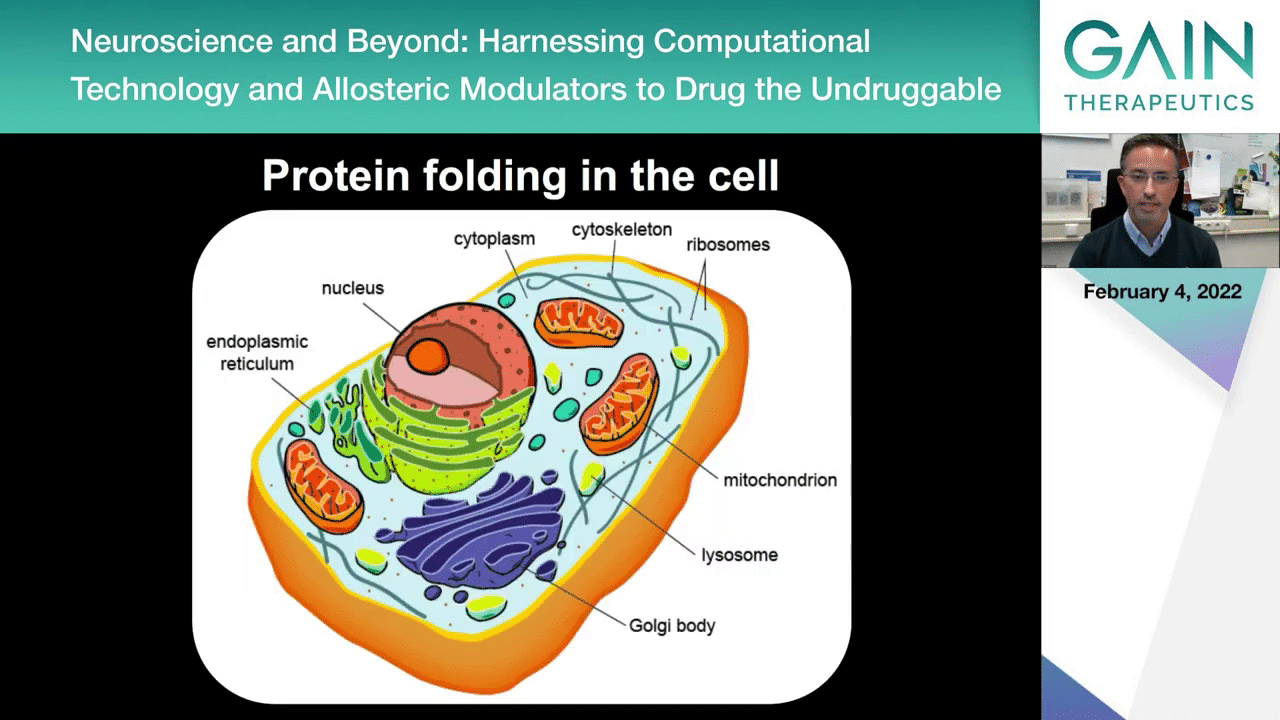
The problem is that when we study the process of protein folding in vitro, if we use simplified systems, we ignore much of what happens in the context of a living cell. This is a textbook example of what a eukaryotic cell looks like and you probably recognize all the components shown here. The nucleus of the cell, the mitochondria, the cytoskeletal elements, even the little dots that represent the ribosomes, but then we don’t have a good way for representing the actual crowded environment of the cell. We tend to represent a cell as if everything was sort of floating around with a lot of space in between the components, but things are not quite like this.
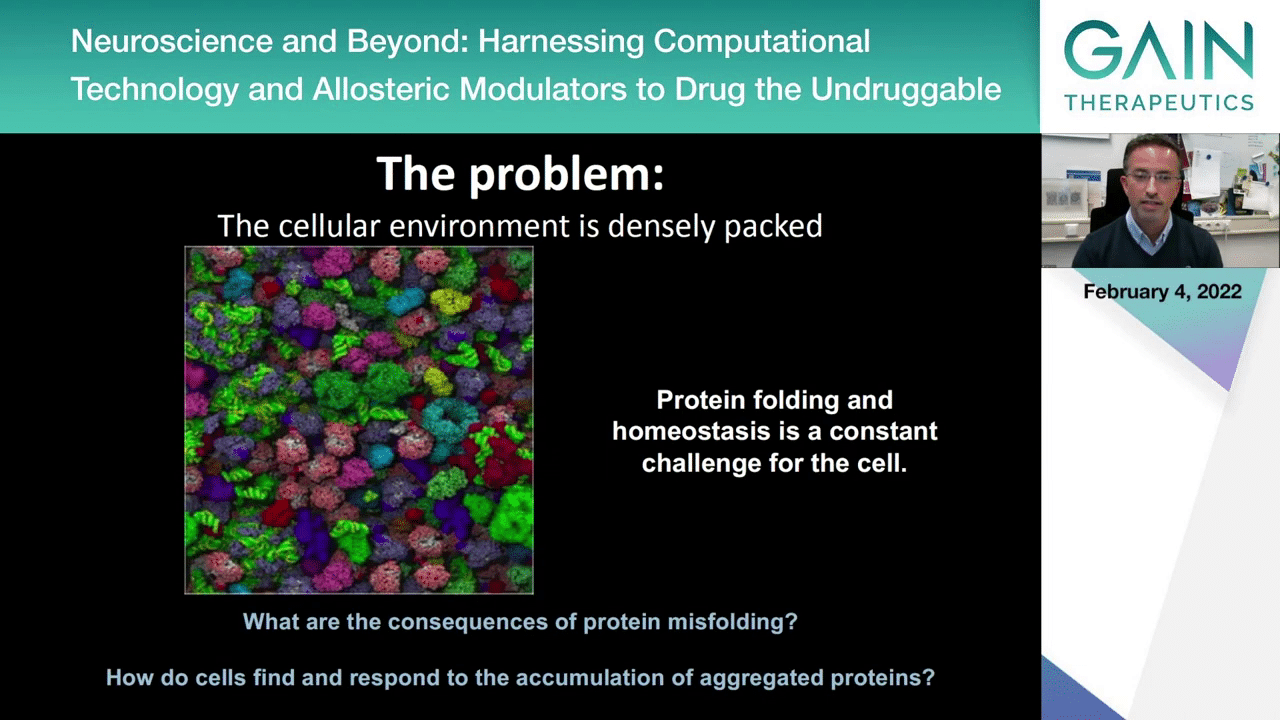
In the next slide you will see something that we think represents better what the intracellular environment looks like, and this shows you various proteins and biomolecules that create the very densely packed env, and this creates many challenges for the cell and for the proteins and other biomolecules as they are synthesized. So, we know actually that that many of the newly synthesized proteins in the cell never become functional because they often encounter problems that prevent them from folding properly, and this requires that the cell has quality control mechanisms to check whether these proteins are folding properly, whether they are functioning properly, or whether they need to be recycled into their components so that new functional proteins can be synthesized.
For many years we’ve been studying these basic questions such as the ones I listed here. What are the consequences of protein misfolding in the context of a cell? How do cells respond to the accumulation of these aggregated proteins?

I like to think of things in simple terms, so here, this is a simple way to represent the process. You can start with a protein that has no structure. Here, just a sheet of paper, and then it undergoes a series of steps that may enable it to acquire a folded structure that allows it to perform a function. But we know that during this process things can go wrong and proteins may not adopt the proper folds that they need to adopt, and they misfold and we know this is not good. This is associated with disease.

We know that on one hand, because you don’t achieve the proper structure, you lose the normal function of the protein and in the context of many of these diseases that we are talking about and neurodegenerative diseases in particular, we tend to think of a gain of function that in this case is a gain of a toxic function because we think that the misfolded proteins may actually cause cytotoxicity in the cells if the cells cannot get rid of these proteins.
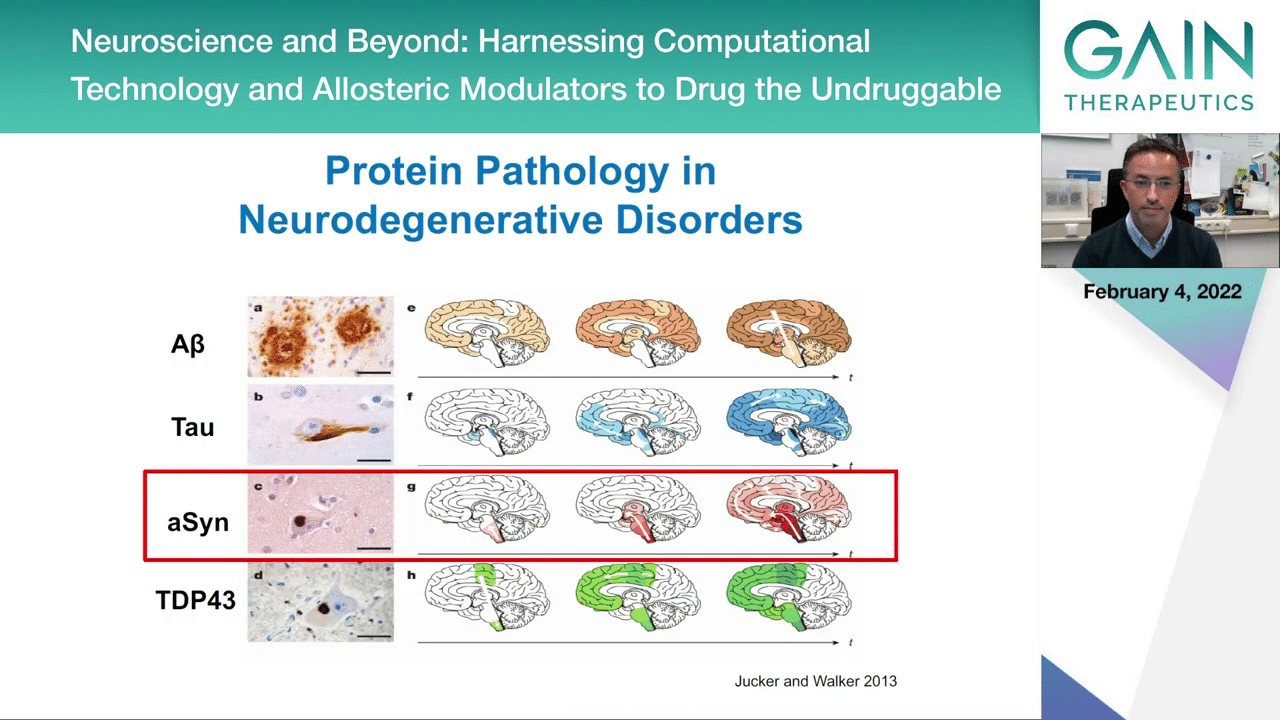
This is actually true in many, if not all, the neurodegenerative diseases that affect the brain, and now we now affect other organs as well. Here you have typical protein accumulations that you find in the brains of people with Alzheimer’s diseases. This A-beta, it’s a peptide that accumulates and forms the A-beta plaques in the brains of patients with Alzheimer’s disease. You see also the Tau neurofibrillary tangles that are also a pathological hallmark in Alzheimer’s disease and other diseases known as tauopathies. Then you have alpha-synuclein forming what we call Lewy body inclusions that accumulate inside neurons in the brains of patients with Parkinson’s disease, dementia with Lewy bodies, and a few other more rare diseases. Then you have also proteins like TDP43 that accumulate in aggregated forms in diseases like amyotrophic lateral sclerosis. I’ll focus in the remainder of my talk on alpha-synuclein, so if you bring the next slide, please.

Alpha-synuclein, as I mentioned, is associated with Parkinson’s disease, with dementia with Lewy bodies, and also multiple system atrophy. It aggregates in slightly different types of aggregates. The aggregates you find in multiple system atrophy, they tend to be a bit different and found in different cells than the ones that you find in the brains of patients with Parkinson’s or dementia with Lewy bodies.
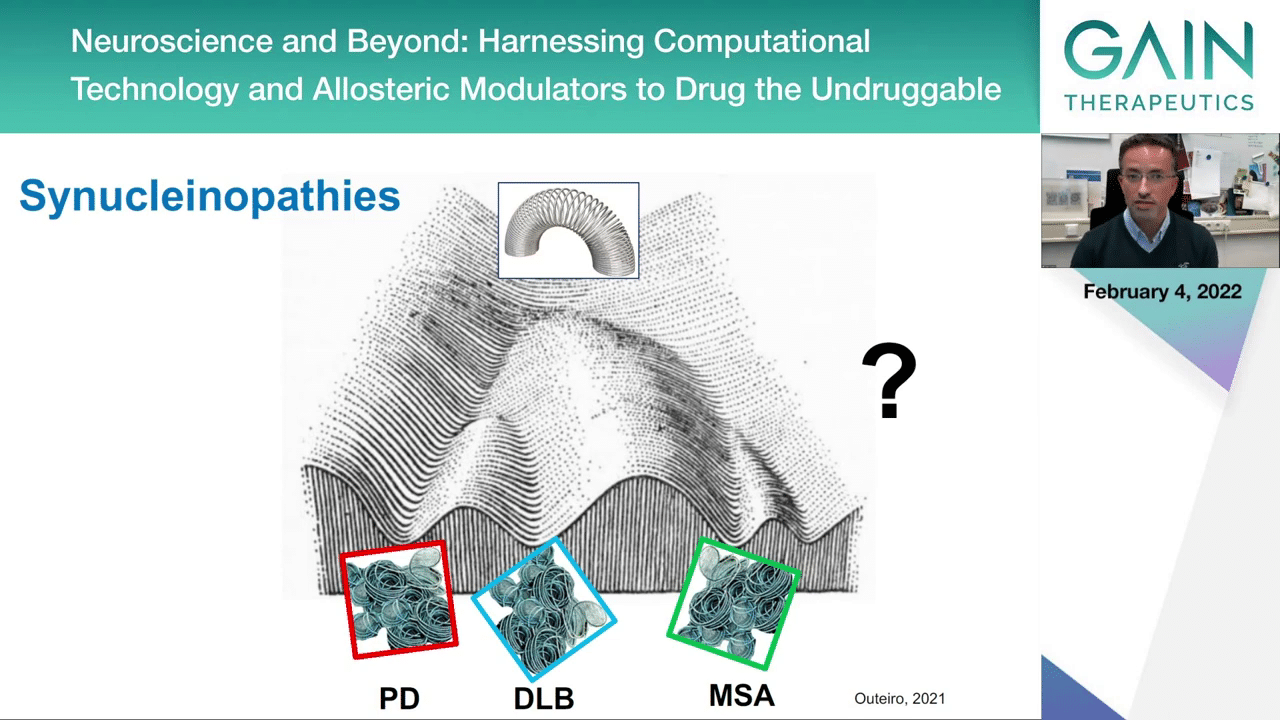
The way we are thinking about this is that you start with the same protein, with a certain shape, and then due to reasons we do not fully understand, you end up with protein accumulations, protein aggregates that look slightly different. This is what new technology like Cryo-EM is starting to show us, that perhaps the different diseases are because the proteins are accumulating with slightly different shapes, and this can lead to different problems in different cell types.
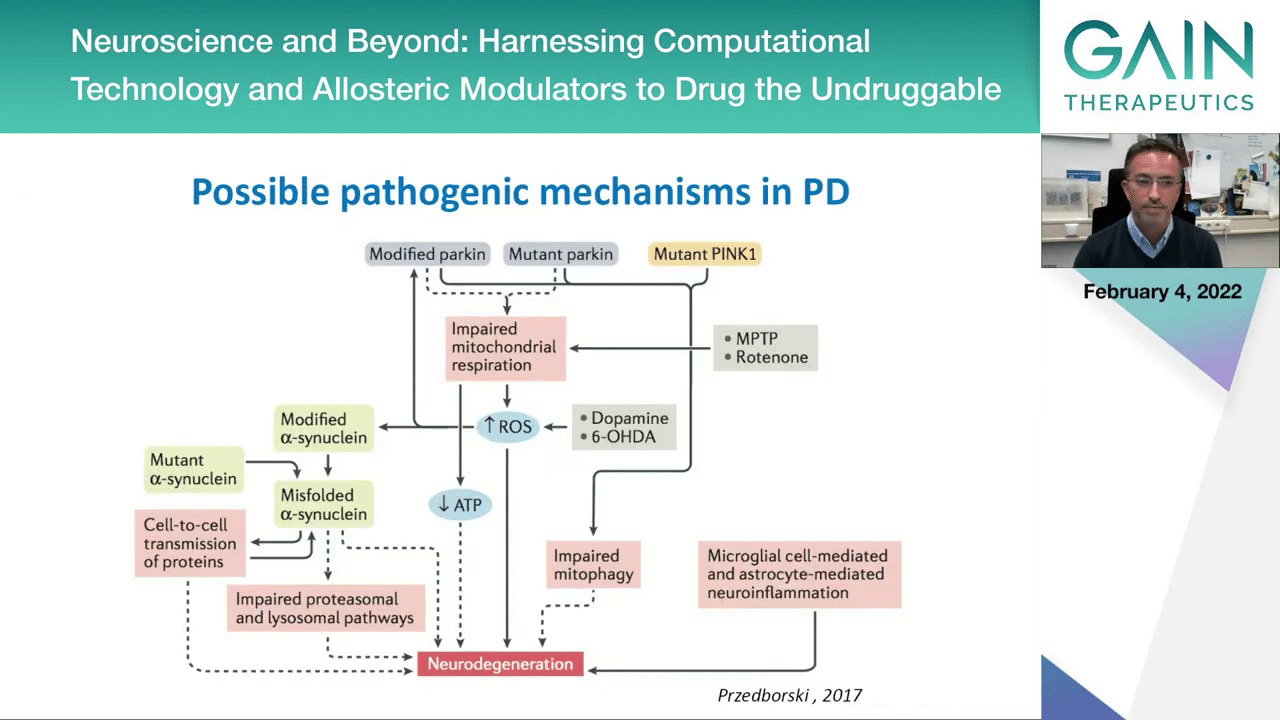
When you think about the possible pathogenic mechanisms in a complex disease like Parkinson’s disease, the picture is very complex, and this is not even the full picture. We know that there would be other pathways and genes to include here, but this is a summary of the pathways and genes that we think can lead to a disease that manifests with the typical symptoms that we normally use to describe Parkinson’s disease.
Here you see that on the left side this protein, alpha-synuclein, is mentioned a lot. We can have mutations in this protein leading to familial forms of disease. You can have certain modifications like the phosphorylation on serine 129 that we think may be associated with pathological forms of alpha-synuclein. Then we have other concepts like the spreading of pathology and the impairment of the quality control mechanisms in the cell that would normally be able to clear and degrade the protein aggregates.
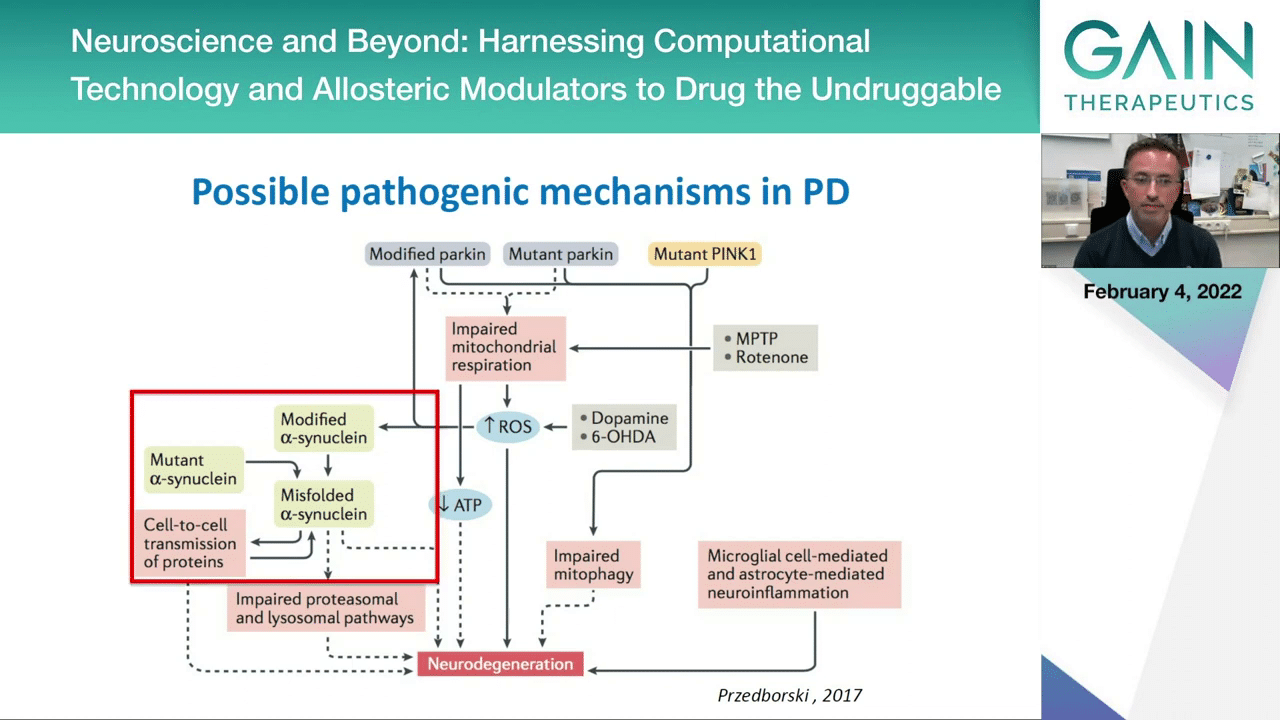
For the remainder of my talk I will focus on these particular aspects of Parkinson’s disease because they are more relevant for what we are talking here.
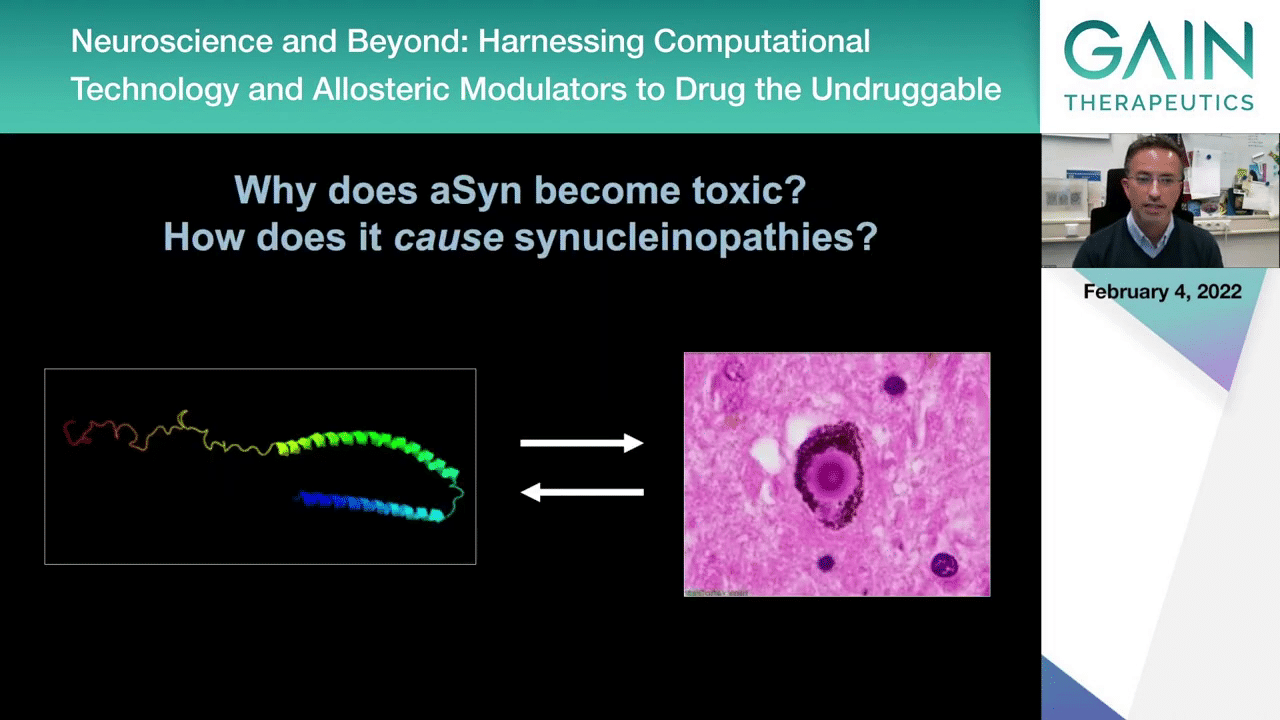
We still need to address these basic questions, why does this protein alpha-synuclein become toxic and how does it cause synucleinopathies? This is intimately related to this idea that you go from a typical normally folded structure into an altered form of the protein that accumulates and deposits inclusions in the brains of patients.
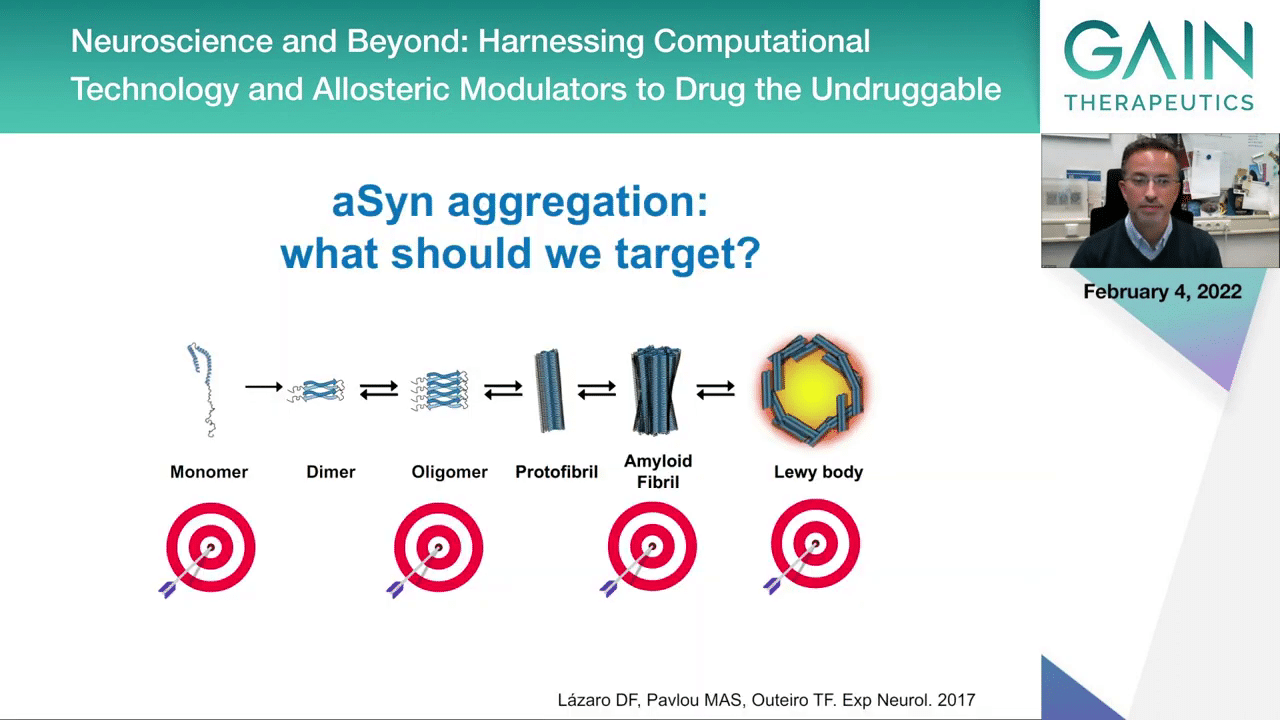
When we think of the aggregation process, this we think gives us ideas of what we should target. Should we target the end product of the process that we think is the Lewy body, the aggregated forms that we can easily see upon pathological examination of the brain, or should we actually try to target other forms of the protein that may accumulate along the way and that may actually be more toxic? We talk a lot about oligomeric species of alpha-synuclein and all of these other proteins associated with other diseases that we think might be more toxic, more reacted with other cellular components than the actual protein aggregate. We still don’t know and we need to test these various possible targets if we want to be successful in terms of therapeutic intervention.
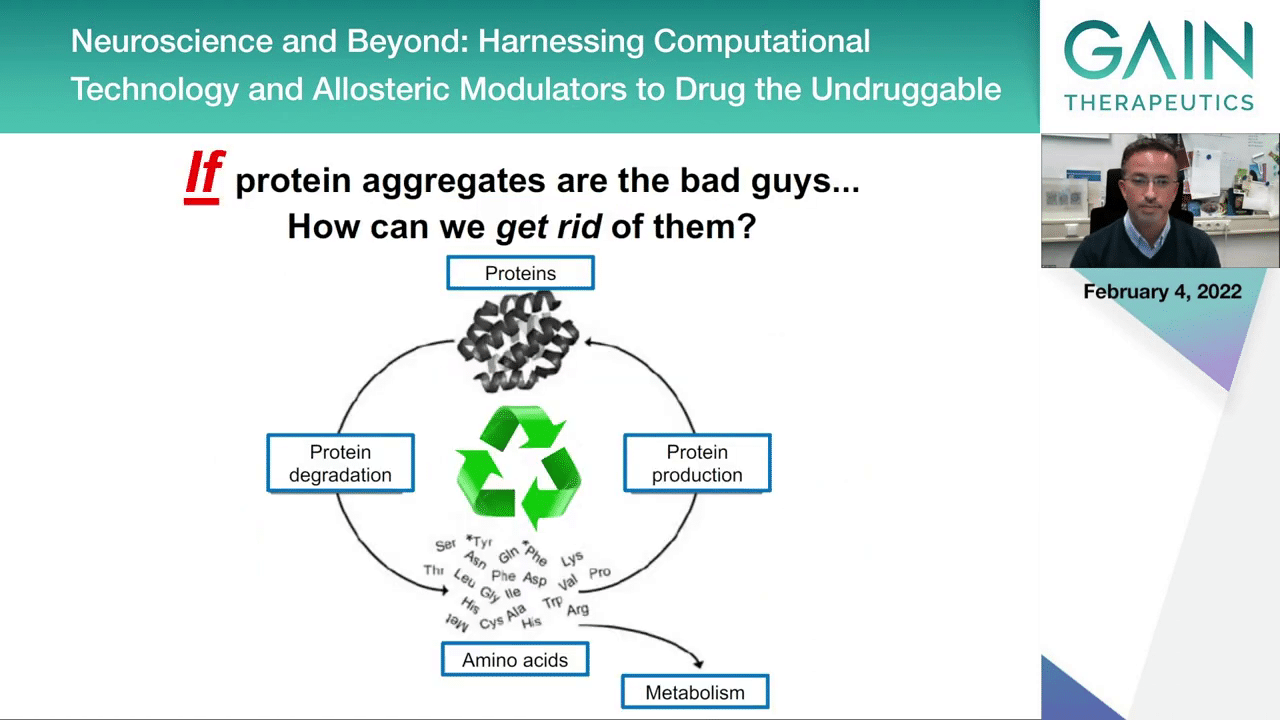
If you think about the aggregates and that these can be the bad guys, so to say, you have two possibilities to interfere with the process.
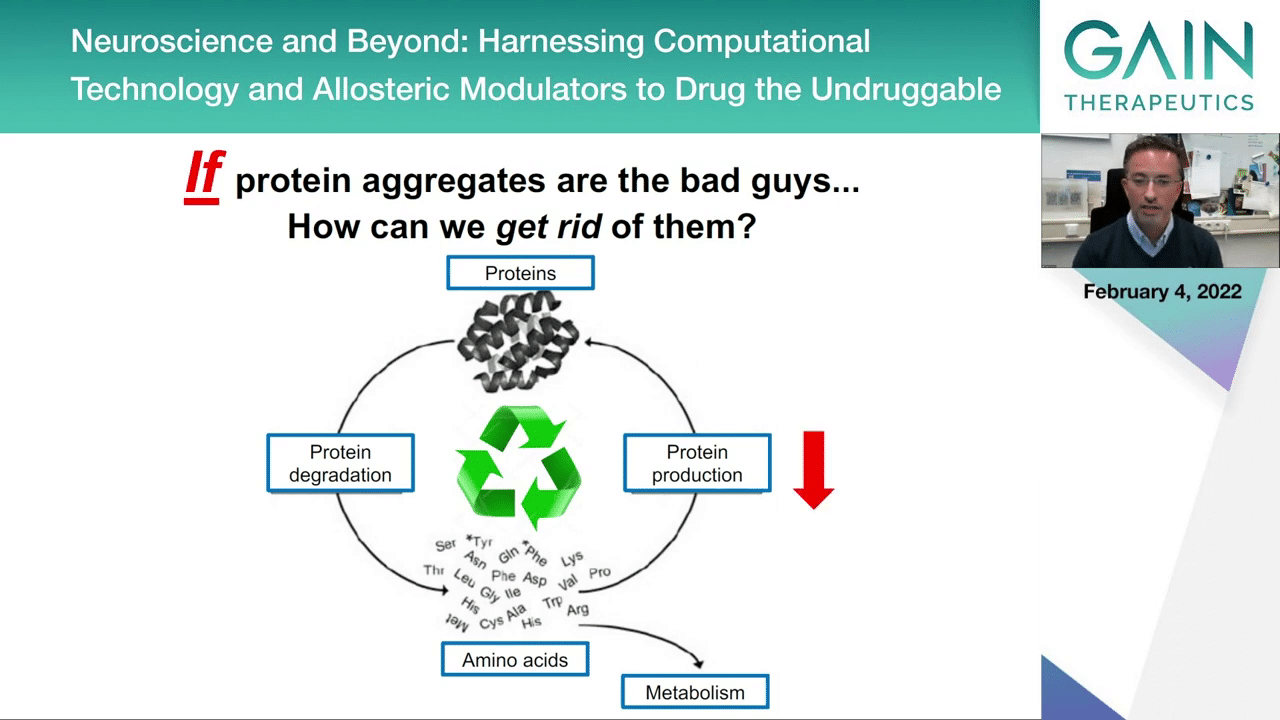
One possibility would be to reduce protein production. This would make sure that if you don’t produce as much protein you will be less likely to accumulate the toxic forms of proteins.
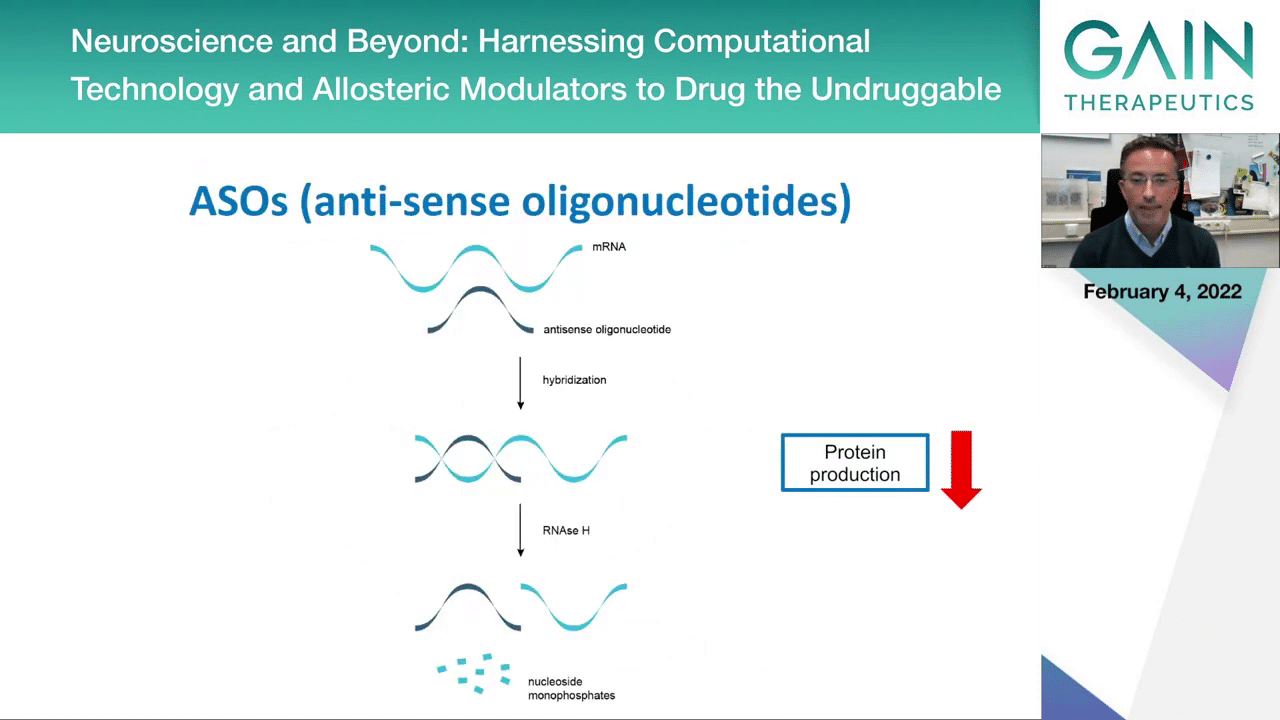
To try to do this, there’s different strategies now being attempted by different companies. One is the use of antisense oligonucleotides that interfere with protein production, so this could be one strategy.
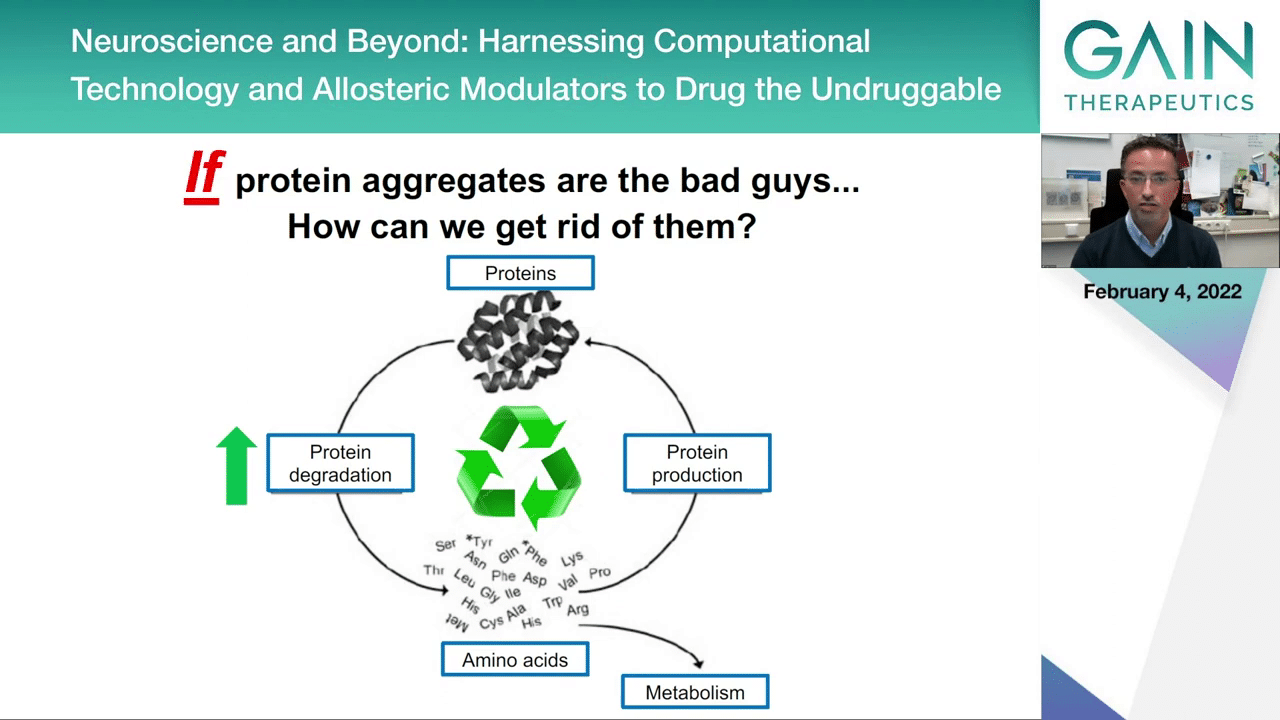
But another strategy could be to try to increase the protein degradation. This would be by exploiting the cellular quality control mechanisms like the ubiquitin-proteome system that we already heard about, and the lysosomal or autophagy lysosomal pathway which is also involved in the degradation of proteins and in particular larger proteins and cellular components that could be accumulating during disease.
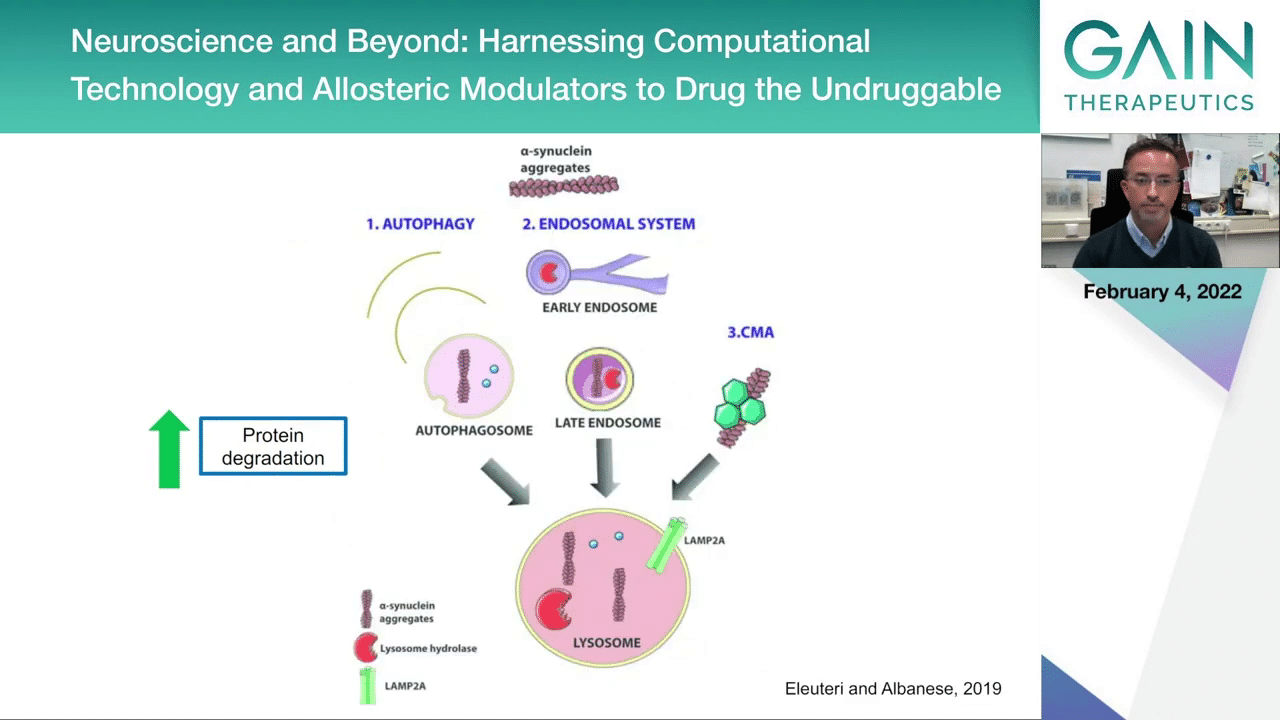
This is a summary of pathways that could be targeted if we want to then achieve protein degradation. I mentioned the lysosomal pathway that would ensure that the aggregates are captured in the compartments inside the cell where various proteases would then be able to access the proteins and the protein aggregates and chop them off so they could be recycled into new proteins. But this is actually not very easy to achieve. There’s several challenges that still haven’t enabled us to be very successful.
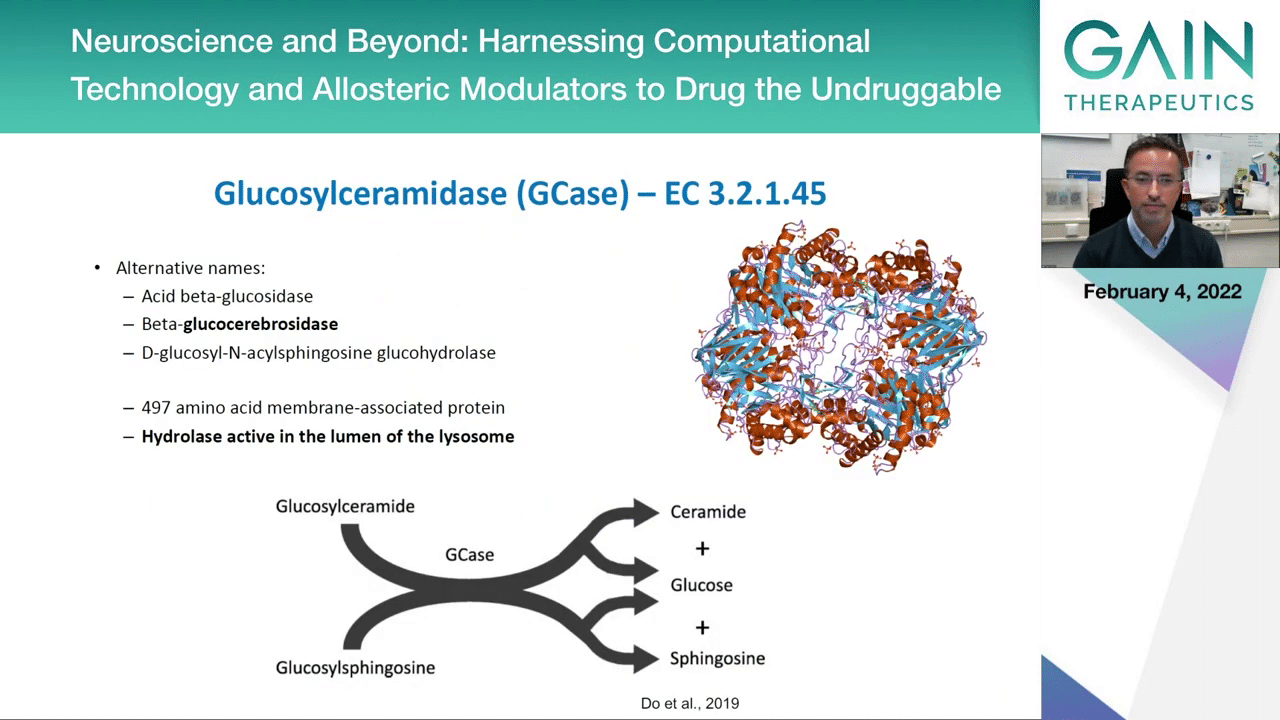
This is where this enzyme that we already heard about, GCase or it has several alternative names; the most common is the beta-glucocerebrosidase. This is an enzyme that is present in this compartment, the lysosome, and is involved in the metabolism of specific types of lipids, sphingolipids, and it converts, for example, glucosylceramide into ceramide or glucosylsphingosine into sphingosine and it releases a glucose molecule.

This enzyme is important because it’s associated with this genetic disease that we just heard about from Manolo before. It’s a lysosomal storage disease where the mutations in the gene that encodes for the enzyme will lead to deficient GCase activity and leads to the accumulation of sphingolipids that we think are toxic and lead to the multiple phenotypes that are typical in this disease.
What was interesting was that when this disease was studied some decades ago, people realized that many patients with Gaucher disease would develop Parkinsonian symptoms.

This connection between GBA mutations and Parkinsonism came to the prime light in Parkinson’s research because in the 90s researchers identified Gaucher patients who developed Parkinson’s disease later on in their lives, and then the GWAS studies that were made actually established that GBA mutations are one of the strongest risk factors for the development of Parkinson’s disease, and these account for 5% to 10% of Parkinson’s disease cases.
There are several features in GBA mutation carriers that are in line with what is observed in Parkinson’s such as the loss of the dopaminergic neurons that produce the neurotransmitter dopamine that is important to control movement, and actually, it’s interesting to see that these mutations are not just associated with Parkinson’s but also with other synucleinopathies, establishing here a connection between this enzyme GCase and alpha-synuclein.
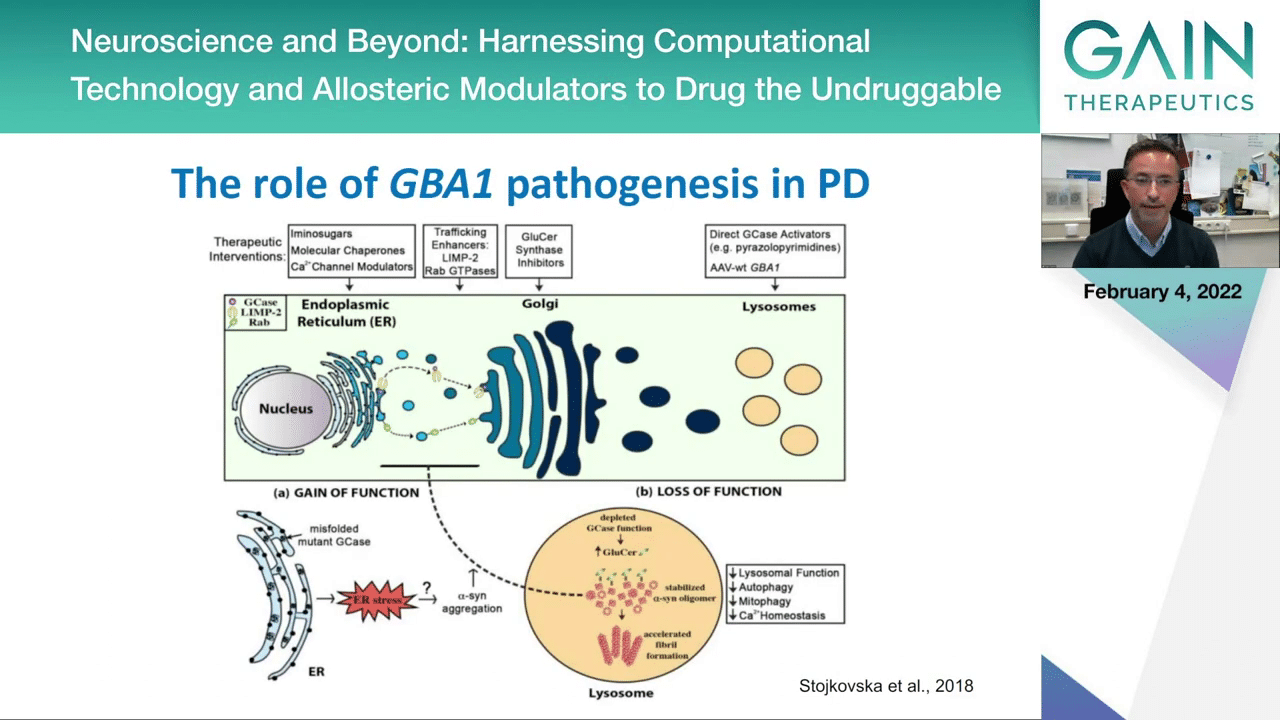
This is how we think it works. We know that this enzyme GCase is produced in the ER, that it traffics and goes through the cell, going to the Golgi and then eventually reaching the lysosome where it should be active in order to degrade alpha-synuclein, for example. But we know that the mutations in GCase can actually cause it to misfold, to never reach the end compartment where it needs to go, and the protein can accumulate then in the cytosol, can lead to different types of stress, and because it will not be properly active in the lysosome then the proteolytic activities will not be functioning properly, it will not be able to degrade alpha-synuclein and these proteins can accumulate, thereby perhaps causing disease.

With this knowledge in mind, we need to step back and look at where we are. At the moment in the context of Parkinson’s disease and other neurodegenerative diseases, clinicians are pretty good at detecting clinical symptoms, and then whenever this exists they can apply available therapies. So in Parkinson’s we actually are in the privileged situation where medication can actually work in the brain circuitry that regulates movement and this can have several beneficial effects for some time in these patients. But the problem is that we don’t understand the causes.

What we need is to understand the causes with a lot of strong and focused big science and this is where the future therapies will come out of. We need to really study processes like protein aggregation. Not only, but also protein aggregation and hopefully come up with targets that we can actually use for improving the lives of the patients.

This is just a summary from a recent paper from colleagues in U.S. and now in Israel where they summarize the strategies that are now being attempted to modulate single lipid metabolism. As you see here, again we are in this idea of having small molecule chaperones, so these allosteric modifiers, and there this is where the molecule that Gain Therapeutics has identified could be playing a big role.
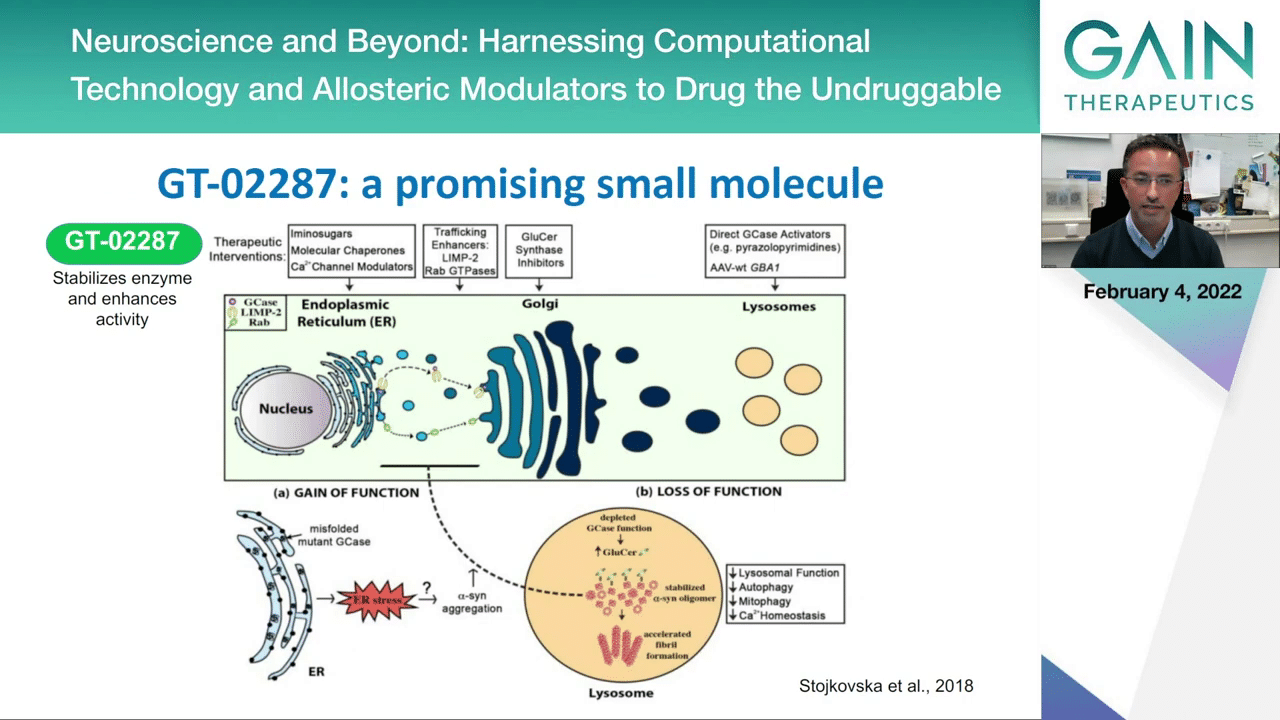
This is, again, going back to the previous schematic, I’m showing you where the molecule could work. We think that it could stabilize the enzyme, and you already saw data showing that it increases the activity of the GCase enzyme, then restoring the end function of the enzyme, which is lysosomal activity and the degradation of the protein that would otherwise accumulate in the brain.
Next slide, please.
I stop here and thank you for your attention.