


Beatriz Calvo-Flores Guzman, presented at the 20th Annual WORLDSymposium discussing preclinical data demonstrating that our clinical-stage GCase regulator provided neuroprotection & restored motor function in Parkinson’s disease models following delayed administration.


Mutations in the GBA1 gene cause a reduction in lysosomal GCase activity, which increases alpha synuclein pathology, a key pathophysiological hallmark of Parkinson disease.
GBA1-PD patients have earlier disease onset, faster progression, and higher rate of cognitive decline compared to non-carriers PD patients.
Therapeutic strategies aiming to restore GCase function are likely to decrease the α-synuclein burden and therefore expected to slow or stop progression of the disease.


GT-02287 is an orally available brain penetrant small molecule, allosteric regulator of GCase. Gt-02287 binds to GCase and promotes its trafficking to the lysosome, where it restores its function.
GT-02287 addresses the main pathophysiological effects on the disease cascade resulting from GCase dysfunction
GT-02287 is currently in phase 1 healthy volunteers and we believe that it has the potential to be a disease modifying therapy for Parkinson’s disease and other neurodegenerative diseases where GCase dysfunction is implicated such as GD, DLB and AD.
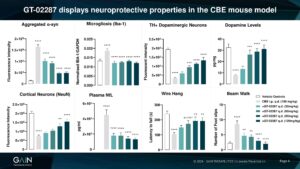

Among the neuroprotective effects displayed by GT-02287 in a mouse GBA1-PD model are the reduction of alpha-synuclein pathology, neuroinflammation as well as dopaminergic and cortical neural survival increase which is reflected in significant reduction of NFL (an emerging biomarker for neurodegeneration)
Amelioration of the pathophysiological cellular events resulting from GCase deficit by GT-02287 treatment leads to motor deficit improvement in the mice.


Gain has investigated the ability of the clinical stage GCase enhancer, GT-02287, to protect dopaminergic neurons and to rescue motor symptoms in delayed treatment paradigms in in vitro and in vivo models of GBA1-Parkinson’s disease.
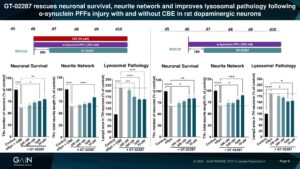

GT-02287 rescued cultured rat dopaminergic neurons injured with α-synuclein PFFs, both with and without GCase activity lowering using CBE, after delayed administration.
Ability to exhibit neuroprotection in dopaminergic neurons injured with PFFs alone suggest that GT-02287 could be a potential disease-modifying therapy for sporadic PD patients that do not harbour a GBA mutation.


Rescue potential of GT-02287 was investigated in a mouse GBA1-PD model, where treatment started either 4 days or 8 days after the initial combinate toxic insult.
Motor function and the neuroprotective effect ( measured as plasma levels of NfL) by delayed administered GT-02287 treatment were evaluated.

At different timepoints of the study, GT-02287 significantly rescued motor function in mice injured with CBE + PFF compared to untreated mice, both when applied either 4 days or 8 days after the initial combined toxic insult.
CBE + PFFs animals treated from day 8 of the experiment had a better performance on day 27 than they did on day 14, suggesting motor deficit reversal with longer GT-02287 treatment duration.


Rescue of motor impairment by GT-02287 treatment is reflected in decreased plasma levels of the neurodegeneration marker neurofilament light chain, compared to untreated mice.
Neuroprotective effect by GT-02287 treatment can be monitored in an accessible body fluid suggesting a great translatability in clinics.


These new data support the potential of GT-02287 as a disease-modifying therapy for Parkinson’s disease that is already clinically established.
Plasma NfL, an emerging biomarker of neurodegeneration, was reduced to control levels in a mouse GBA1-PD model, reflecting the motor deficit rescue observed in the wire hang test.
GT-02287 appeared to reverse motor deficit as treatment continued.


