


Speaking at the Gain Therapeutics R&D Day, Dr. Xavier Barril, shared about discovering Novel Allosteric Targets by Combining Advances in Big Data and Computational Technology. You can watch his entire presented below:

Here is the Transcript of the Presentation by Dr. Xavier Barril
Thank you, Joanne. Thank you very much. We can go to the next slide, please.


I think this is well known that there are many proteins that are associated with diseases somehow, but we haven’t been able to still use those opportunities to develop therapies. It’s because many of them are just difficult to handle with a terp-like molecule. Really, the allosteric drug is (inaudible) is aiming to transform that, as has been said before.
What we have is a tool to do that, which we have (inaudible) as you will see. What we have is a method based on molecule simulations and physics that could provide quantitative predictions of binding hot spots and from that we can discover molecules using supercomputers and the information about the structure of the protein.
This is something that we have been developing for a number of years, so the technology is fairly mature, is validated, and still improving because technology is growing so quickly. We always have new features.
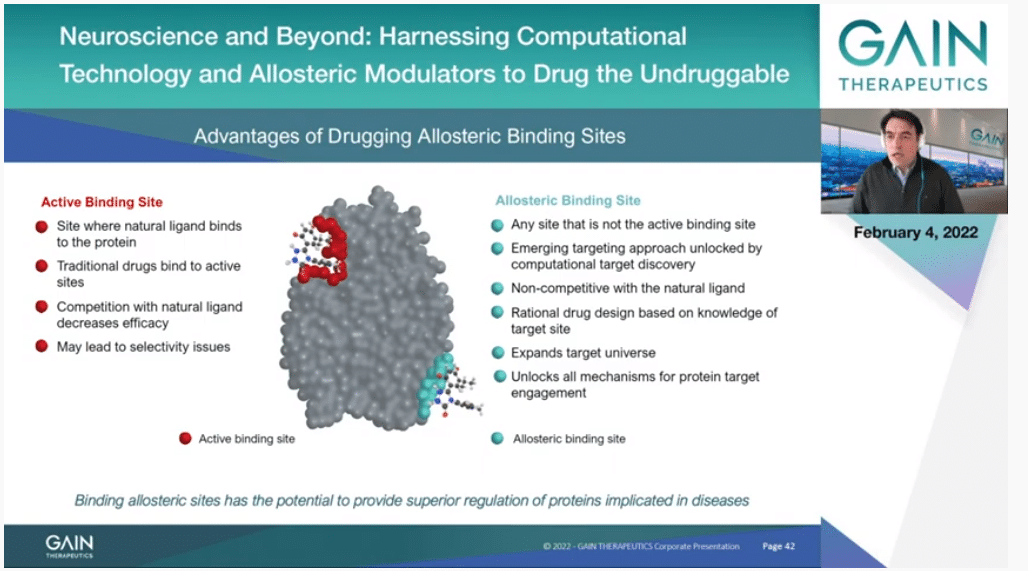

If we go to the next slide, this is I think all redundant with what we have seen, thanks to the introductions by Dr. Cournia and Dr. Broder. But just to insist that this is what we faced in the very beginning and the early days of the Company. We didn’t want to fit the orthosteric binding site even though our targets had their own sites because we knew that it could give us an advantage if we could avoid competing with substrate. We could then find new properties and new molecules had completely different properties.
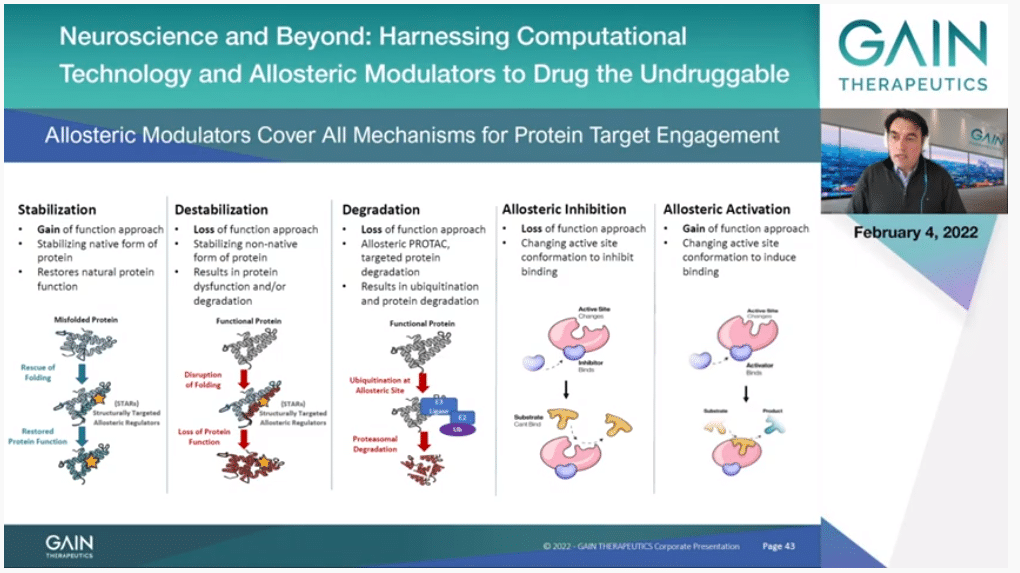

If you go to the next slide, this is just a picture of the different things that you can do by targeting to allosteric binding site instead of the orthosteric. You can sterilize the protein. This is just by binding to the protein in the allosteric binding site, you make it more stable, and you can have a gain of function. That’s where the Company’s name comes from originally. That’s been our primary objective for some years. But we have actually now expanded that. You can also destabilize in the same way by targeting a confirmation that is not the native confirmation of the protein. A particular form of that is actually—and this is a really exciting development. I guess most of you have heard about targeted protein degradation with the PROTAC approach. The PROTAC process needs a molecule that binds to a protein, it’s like a handle to the protein. It doesn’t have to touch anything that is functional. So, any protein that doesn’t have an active site, if you can find an allosteric binding site and a molecule that binds to it, you can destroy the protein. Not just inhibit it as before, you can completely destroy it, and this has a very profound effect.
Then we have the classical allosterism that Professor Cournia presented. When you look at systems in vitro you can see that you can inhibit and actually using the same site you can activate as well. This is wonderful because it provides a huge variety of more subtle ways also of moderating your target than just an orthosteric drug just goes there, inhibits and makes the protein inactive. This is a broader range of function that you can achieve. That’s what we are now looking at, at the whole picture.
If you go to the next slide, please.
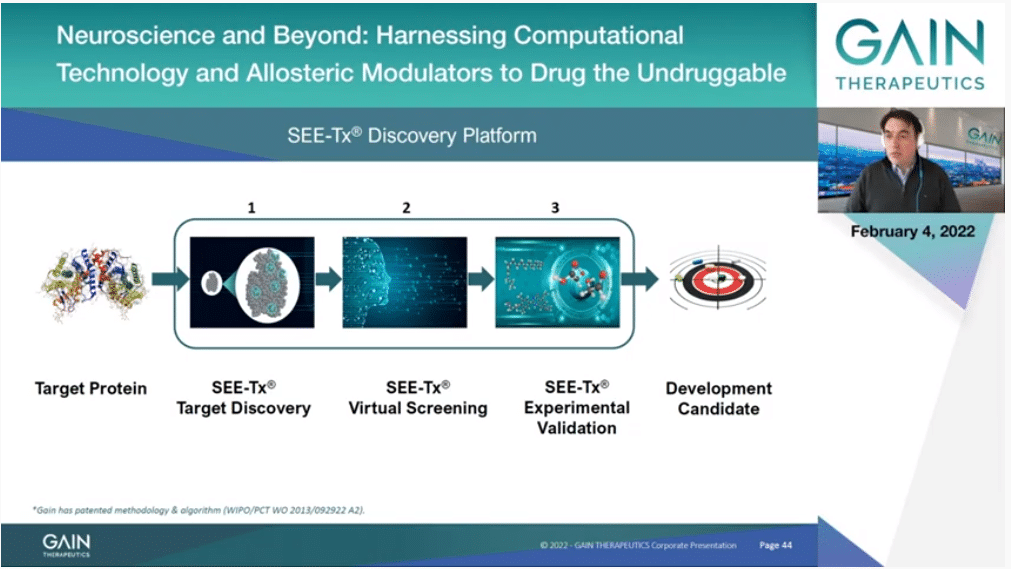

How do we do that? We need the structure of the protein, and thanks to the incredible advancement in structural biology and more recently in computation and AI and the AlphaFold program, that’s essentially the whole of the human proteome, so almost any protein we can work on.
Then of course we have to discover the binding site, then we discover molecules that bind to it, then we have to, of course, do experiments and validate that that’s the case, and then we have to develop those molecules into candidates. Let’s see each one of those three steps a little bit more in detail.
If we go to the next slide, please, it’s actually a movie.
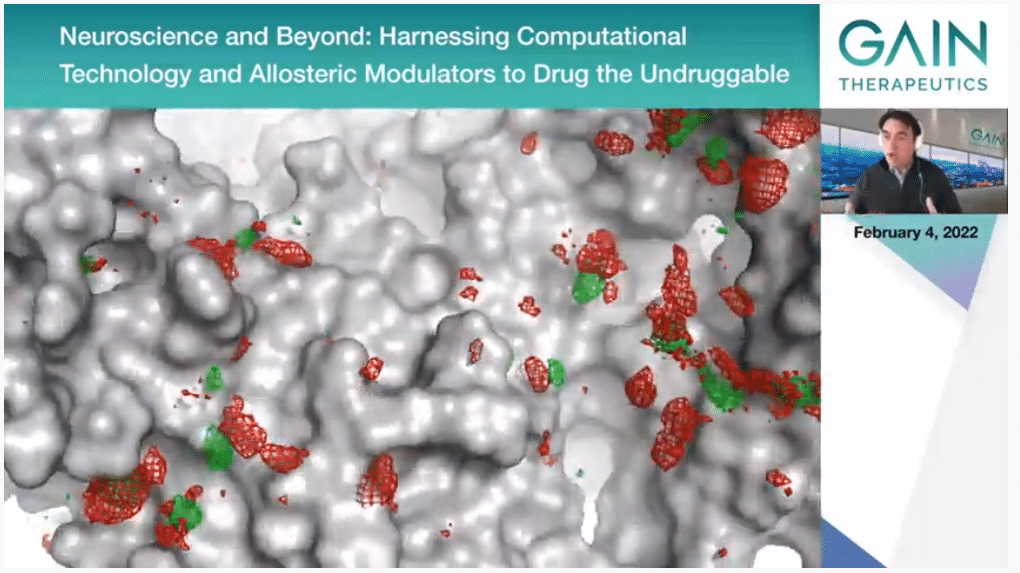
This is just to provide more of a visual expression of what we are doing actually. The core of the technology is the molecular dynamic simulations where in the supercomputer we simulate the protein that is moving, and it is actually interacting with water molecules and with organic solvents. We can simulate this for a relatively long time. Then we can see—sorry, I’m seeing another movie now. I don’t know if you’re still on the first one. Sorry about that.
What we can see is if the protein prefers to interact with water or with organic molecules. This, which is obviously very visual, what we are showing actually, we have complex algorithms that analyze this and extract from that binding free energies that identify the most important binding points on the protein surface.
If you go to Slide 46, yes. No, the previous one, 46. Okay, this one.
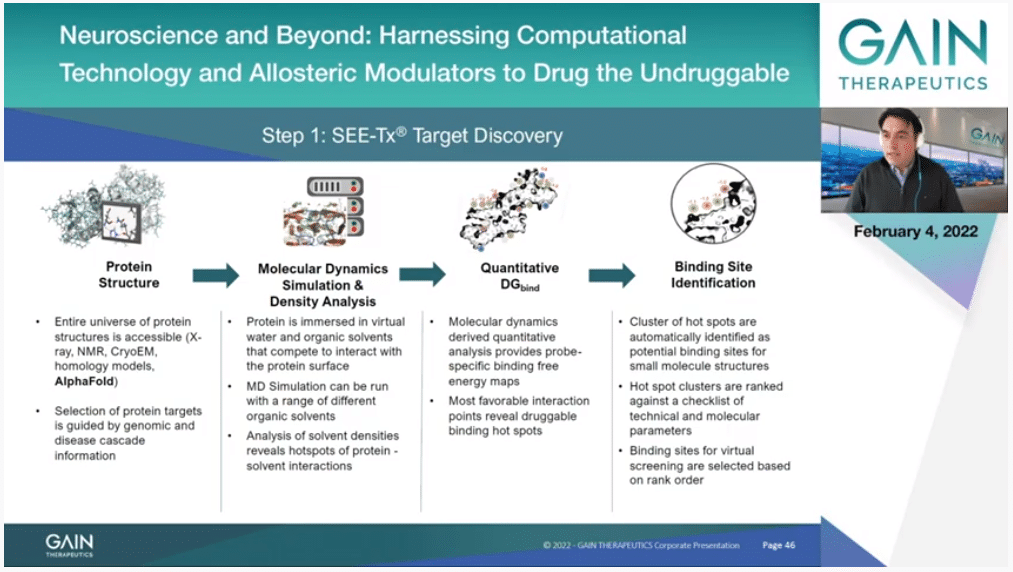

This is a bit more detailed explanation of exactly what we do. We start with a structure of the protein and as I said, the source it can be anything. It could be homology modeling. It can be an AlphaFold model. It can be express (phon) structure or (inaudible) model. Then we have to obviously analyze and check that everything is properly—there is nothing wrong there. Then we can run the simulation that you were seeing before and extract this essential information about binding hot spots. Importantly then we have a third step where we apply our algorithms that are proprietary and subject to a patent, where we actually quantify the exact binding free energy that you can get by placing a particular atom type on each point of the protein surface.
Then, finally, we have a clustering algorithm that identifies allosteric binding sites as a collection of binding hot spots.
Our definition doesn’t rely on shape, it doesn’t rely on anything else on this physics based simulation, and tell us quantitatively if you have a binding site that is druggable. This is quite unique and opens the door then to identify molecules that also bind to this, and this is the next step.
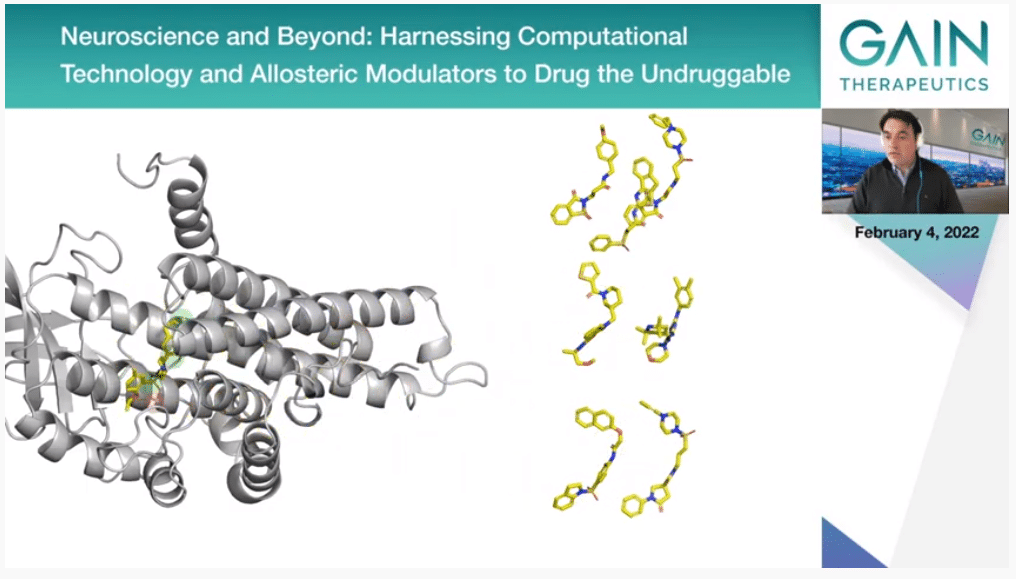

Now you should be playing the second video, where you will see here the binding site that we have identified in this previous step. The dots there represent the key binding hot spots that we have also identified, and then we screen millions of compounds and soon we will be working with actually billions of compounds that we match against the protein structure. Particularly, we look at which ones of them can fulfill the binding hot spots we have identified before.
So, we end up selecting a very small subset of these molecules. You can stop the movie now and go to Slide 48. Thank you. These are the ones that then are going to be selected.
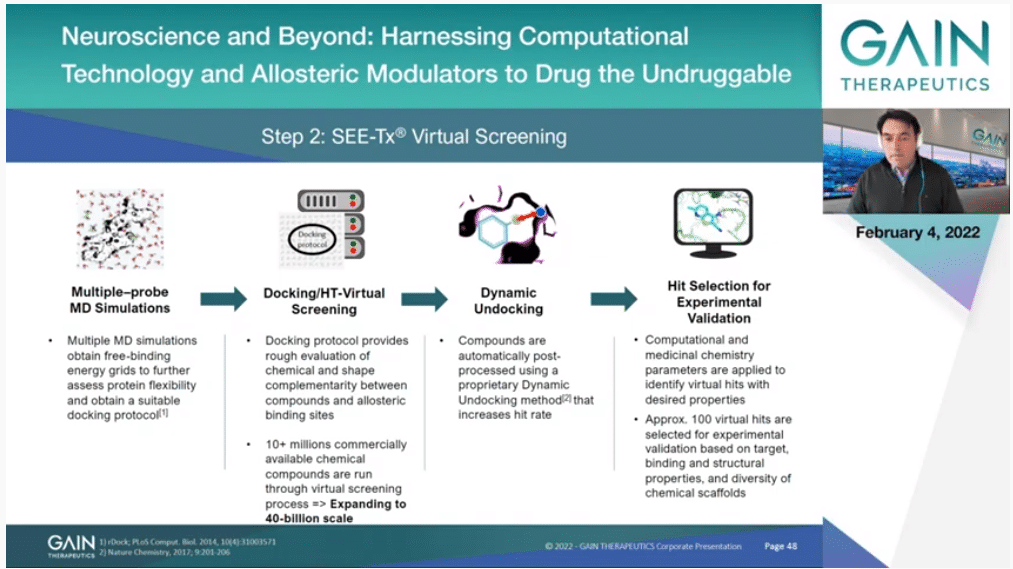

The process is a bit more convoluted. The movie can give you an idea of how this works, but it’s more convoluted. We look at different atom types that can be—how they interact. We can look at the flexibility of the protein in more detail. We are also using other methods after docking to improve the results, methods that we have developed by the way. Finally, what is important, we ended up selecting a very small set of molecules, just in the order of 100 or so, that then they will be experimentally tested.


This takes us to the next slide, and just with the multiple projects that we have done in the Company over the years, the average time that it takes us to decide if an allosteric—if a protein contains a druggable allosteric site is one or two weeks normally, so that is very, very quick. Then the virtual screening of about 10 million molecules is less than one month.
So, actually the majority of the time is just setting up the assay and testing those compounds that we select. On average for binding sites, and remember these are always binding sites that never have been targeted before because people did not even know that they existed. We have had 14% of success. That means that if buy 100 molecules we have 14 that are active, which is plenty to start heat (inaudible) and then the evolution of those compounds, and start drug (inaudible). That’s at the very least more than 1,000-fold better than just from doing the random screening by high throughput screening, for instance.
Then if we go to the next slide, this is very important.
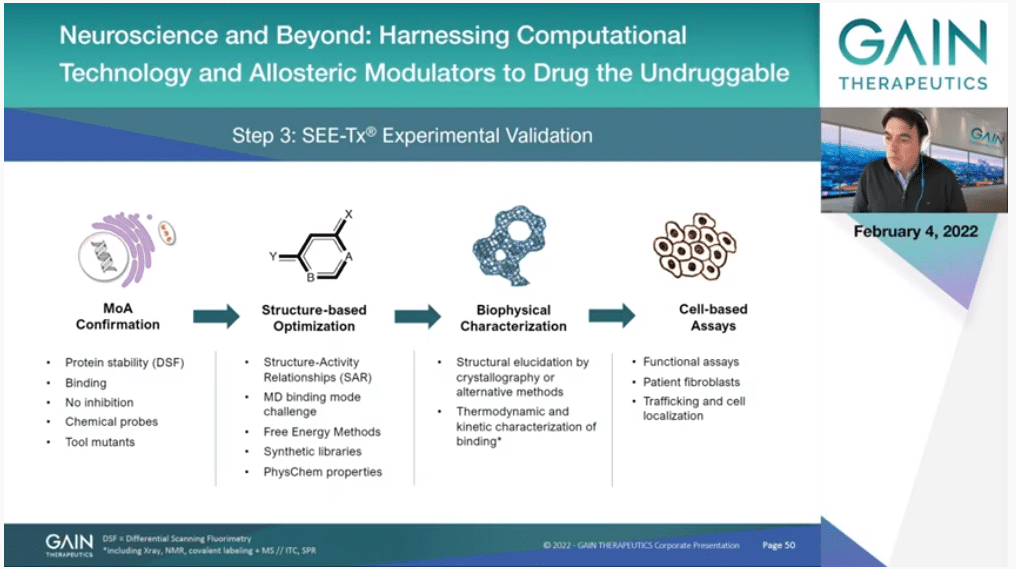

We have obviously emphasized the computational technology because this is something that is quite unique, but the Company is very much real; it’s not virtual. Once we have compounds that are active, we have the drug discovery process and look at the science in detail, understand the mechanism of action with biophysical techniques. We have to do SAR with medicinal chemistry, understand how we can improve the properties of the molecules. We support that with computational methods as much as we can, but of course there is medicinal chemistry hardcore analysis of what’s happening and improving those molecules. And structural biology, of course, and in the end biology is king here so we have to follow always a biological signal. We have to have a therapeutic effect. So it’s not just a matter of binding.
This is what I want to emphasize on the next slide is that allostery, of course always computational has to go hand-in-hand with experiments, but in allosteric drug discovery this is particularly true.
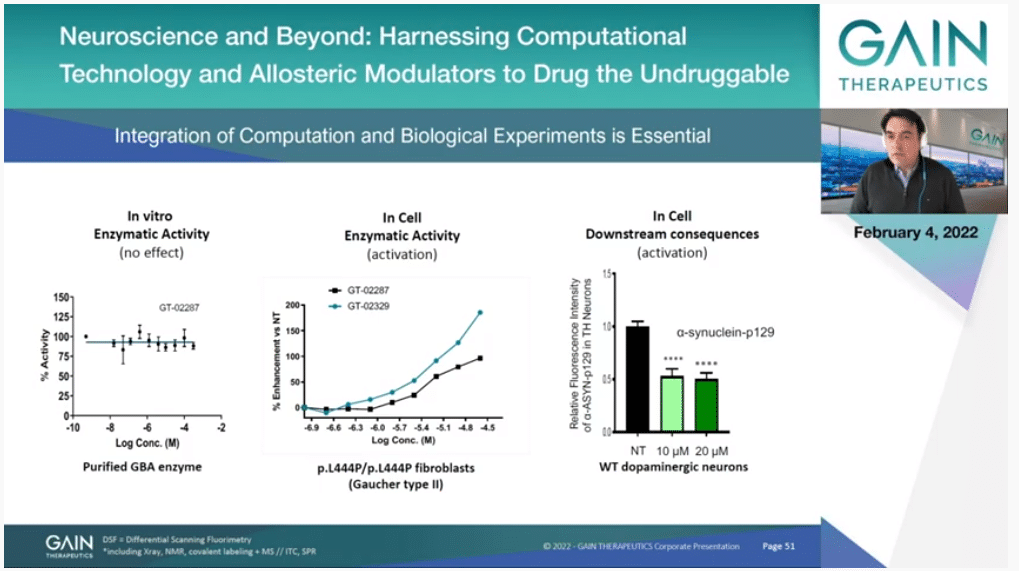

If you look at the first panel to the left, this is one of our molecules in an enzymatic activity assay. The flat line means it’s doing nothing. It’s a silent binder. We know that it binds because we have other means, other experiments that I’m not showing but we know it’s binding, but it’s not doing anything. If you look at it just for the protein in isolation. It’s binding elsewhere. It’s allosteric in that sense, but it doesn’t have an impact on the enzymatic activity.
But what happens in the cell? That’s what you have in the second box in the middle, is actually you can see a dose-dependent increase of enzymatic activity. That’s because this molecule is binding to the protein, it’s making it more stable. It’s actually increasing the amount of protein that you have—in this case it’s in the lysosome—and therefore you have more function, more effect. You actually double the amount of enzymatic activity.
Of course, the time is different, so this is an experiment that is over a period of several days. It’s in the right biological context and so on, and then the most important thing is you have to look at the downstream consequences of that, if you are having some consequence that can have a therapeutic effect. In this case, and you’ll see more of that later, we managed to reduce the amount of alpha-synuclein which is a protein that is the root of the pathology of Parkinson’s.
This is a message that is really, really important. In allosteric drug discovery the effect is a lot more subtle. You cannot skip at all the biological part. It’s a key component of what we do.
With that, I come to the conclusion, so next slide, please.

Just to reiterate that SEE-Tx makes allosteric drug discovery fast, efficient and rational. This has been given rise to actually a range of projects, both internal and with collaborators, and Manolo will be showing some of them in the next presentation. As a result, SEE-Tx has been thoroughly validated. This is transformative because as Dr. Cournia and Dr. Broder pointed out before, allosteric drug discovery has huge potential, but the (inaudible) discovery of allosteric drugs is just not accessible and we are pioneers in this field.
We continue to improve the technology, adding new features and capabilities to find allosteric drugs even faster and more efficiently. There are terrific advances in almost all aspects of science and things like incredible growth of chemical space, which is now reaching this 40 billion space that you can buy very quickly and for a low cost, and of course, artificial intelligence, not only on the structural prediction. AlphaFold, of course, is a primary example of artificial intelligence, but also to predict toxicity of targets, PK properties, etc., this is a huge help in drug discovery.
Computation of course is essential to reach successful allosteric drug discovery. But it is also essential to realize that allosteric drugs moderate biology in ways that are not obvious, and the discovery of allosteric drugs must go hand-in-hand with relevant biological experiments.
The biology team, the chemistry team, the pharmacology team are just as important for Gain as the computation or the structural biology, and we are very proud of what we have achieved together. Thank you very much.
